
-
 1自組裝奈米粒子應用於樹突狀細胞免疫強化策略寶泓生醫股份有限公司/ 生物技術發展處許維喆處長發明人:許維喆、林峙諳 、趙敏涵 、李存詔、賴緒明 、白金凌領域:醫療器材, 劑型開發適應症:樹突狀細胞免疫強化研發階段:醫材雛型開發Prototype development摘要:
1自組裝奈米粒子應用於樹突狀細胞免疫強化策略寶泓生醫股份有限公司/ 生物技術發展處許維喆處長發明人:許維喆、林峙諳 、趙敏涵 、李存詔、賴緒明 、白金凌領域:醫療器材, 劑型開發適應症:樹突狀細胞免疫強化研發階段:醫材雛型開發Prototype development摘要:寶泓生醫源自國立中興大學化學系賴秉杉實驗室,透過教育部的新創研發服務公司孕育計畫逐步發展,我們獲經濟部工業局認定,並在中興大學管理學院的支持下成功通過創櫃版創新創意審查推薦。我們的核心技術聚焦於高分子接枝、奈米微胞傳輸以及脂質成分包覆。在樹突細胞刺激表達技術領域中,我們是少數使用奈米顆粒策略的團隊,利用這獨特策略研發出以自組裝奈米顆粒為基礎的AJ系列細胞製劑,不僅成功地強化了樹突狀細胞活化免疫反應能力,更能大幅提升樹突狀細胞的移行能力及抗原呈現能力。目前,AJ系列的技術專利正處於審核階段,而我們堅信這系列產品之投入能在細胞製劑市場中解決自體樹突狀細胞治療的患者個體差異所導致的療效不彰之問題。
Powin Biomedical Co., Ltd. originated from the Lai Ping-Shan Laboratory of the Department of Chemistry at National Chung Hsing University. Through the incubation program of the Ministry of Education's Startup Research Service Corporation, our company has gradually evolved and earned recognition from the Industrial Development Bureau of the Ministry of Economic Affairs. With the endorsement of the College of Management at National Chung Hsing University, we successfully passed the recommendation for the OTC Innovation and Creativity Review. Our core technologies revolve around polymer grafting, micellar transduction, and lipid component encapsulation. In the field of dendritic cell stimulation and expression techniques, we stand out as one of the few teams utilizing a nanoparticle strategy. Harnessing this unique approach, we've developed the AJ series of cell preparations based on self-assembled nanoparticles. These not only significantly enhance dendritic cells (DCs) stimulation but also considerably improve cell migration and antigen presentation capabilities. Currently, the technical patents for the AJ series are under review. We firmly believe that this series will solve low therapy outcome of DCs cell therapy due to immune function deficient of patients and then to bring substantial advancements to the cell preparation market.
-
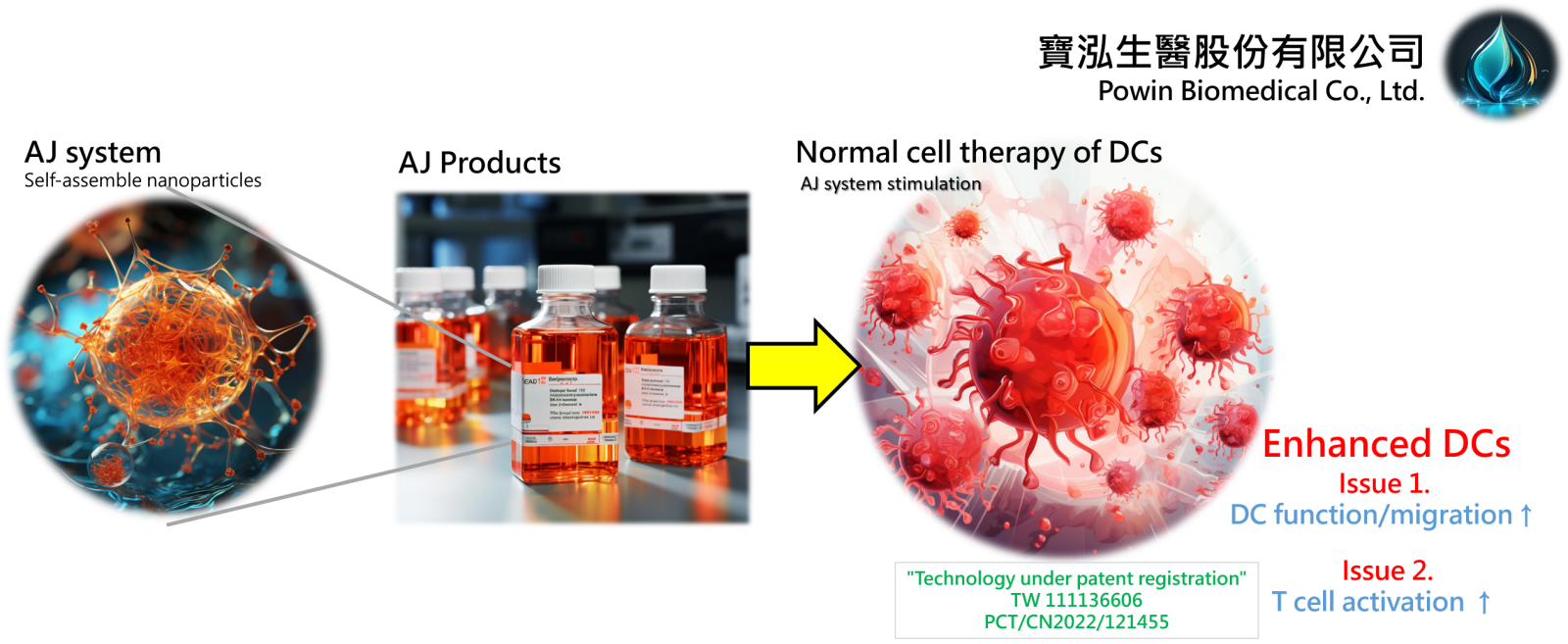
The AJ System employs self-assemble nanoparticles to revolutionize dendritic cell (DC) therapy, overcoming its clinical limitations. This technology enhances DC function, migration, and T cell activation, and is currently under patent registration.
2開發促進腦傷後神經再生藥物國立清華大學陳令儀教授 Professor發明人:陳令儀、王翊、廖彣玲、李育棠、呂亭萱、黃郁文領域:新穎藥物適應症:腦外傷造成的行動功能受損研發階段:動物驗證 In vivo validation摘要:全世界每年約7000萬腦外傷病患,每年花費4500億的醫療成本照護腦外傷病患,突顯了治療腦部損傷的急迫性。目前臨床上治療主要是止血、除血塊、施予預防性抗癲癇藥及3-8個月復健協助,無藥物可供積極促進腦神經再生及功能回復治療。創傷性腦損傷根據其發生的嚴重程度、時程長短與及時就醫與否可能造成不同程度行動功能、語言、情緒、記憶、認知功能的缺失,腦傷病患也是神經退化等疾病的高風險群。受損的腦神經細胞自我修復再生的能力低,後續腦神經再生能力的好壞亦是評估預後良好與否的重要指標。本合作團隊實驗室建立了一套腦神經受傷方法來篩選能促進神經軸再生的藥並研究其修復機制。本團隊已找到能促進腦傷後神經再生的小分子化合物,這些小分子化合物可改善腦傷後行動能力、行為協調能力及大幅減少焦慮表現。國際優先權及台灣專利已提出。團隊成員也正合成化合物及其衍生物,並委外完成該前導化合物的克級合成。研究指出,曾遭受腦創傷的人未來罹患神經退化性疾病的機會高2倍,所以,若能於腦傷後在急性期給藥,促進神經軸再生,可大幅減少未來腦外傷病患罹患神經退化性疾病。除針對腦外傷病患,本團隊開發的藥物,未來或許可應用於對腦中風、腦部癌症放射線治療後產生之神經損傷或神經退化性疾病患者。
Approximately 70 million people sustain a traumatic brain injury (TBI) each year spending 450 billion USD of medical expense. Current clinical care for TBI patients includes hemostasis, remove clots, administering anti-epilepsy drugs as a preventive measure, followed by 3-8 months of rehabilitation if necessary. Thus far, no medicine is available to promote neurite re-growth of injured brain neurons and their functional recovery. Depending on the severity, time before medical treatment and the region injured, patients could express different symptoms, including motor function deficit, language, emotion, memory and cognitive dysfunction. TBI patients are also high risk for developing into neurodegenerative diseases. Due to the limited regenerative potential of central nervous system, promoting regeneration at acute phase of TBI could promote functional recovery and prevent deterioration that may lead to neurodegenerative diseases. Our team has identified a number of small molecule compounds that can promote neurite re-growth required for subsequent functional recovery. PCT and Taiwan patents have been filed for these compounds. We are synthesizing NTHU-3 to a 3 g scale to advance pre-clinical study. As TBI patients are at twice of risk to develop into neurodegeneration, with our drug development, not only TBI patients could have earlier recovery, disease-caused TBI may also benefit from it.
-
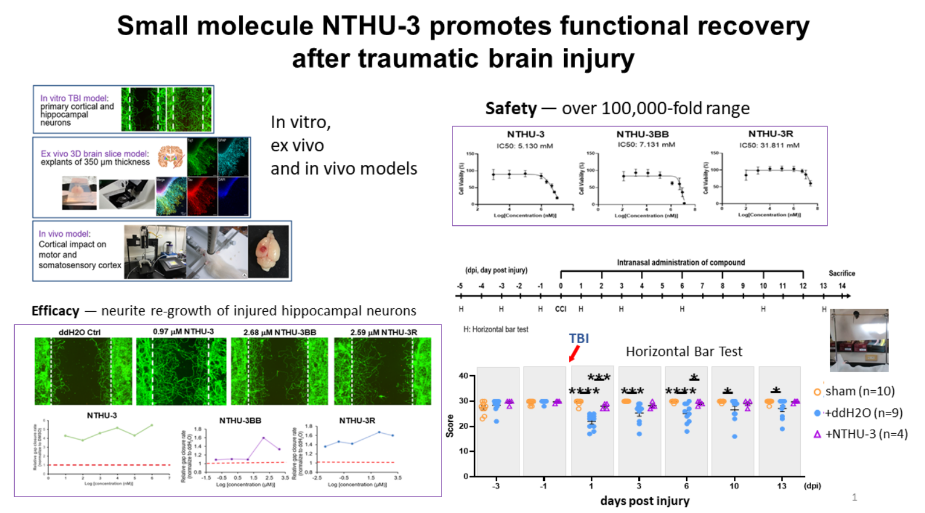
Our team used rat brain neurons (cortical or hippocampal neurons) to screen for compounds that would promote neurite re-growth of injured brain neurons. Safety of the compounds, NTHU-3 and its analogs NTHU-3BB, NTHU-3R, was determined using CellTiter-Glo cell viability assays. In vivo brain injury model used controlled cortical impact to damage mice brain at somatosensory region. Various behavior tests were performed to determine the drug effect on movement and anxiety. The recovery of motor function coordination was determined using horizontal bar test etc.
3治療神經退化性疾病的新策略中央研究院生醫轉譯研究中心陳儀莊特聘研究員發明人:方俊民、林雲蓮、林榮信、林君榮、陳儀莊、黃乃瑰、王鴻利、杜邦憲、陳志成領域:新穎藥物適應症:Alzheimer’s disease, Alzheimer’s disease-associated disorder, neurodegenerative diseases研發階段:申請試驗用藥物/新醫材(試劑) Investigational New Drug (IND)/ Investigational Device Exemption (IDE) application摘要:阿茲海默症是高齡化社會中最常見的神經退化性疾病,隨著全球人口老化,患者已達數千萬人,成為日益嚴峻的醫療負擔與挑戰。現有臨床藥物僅能緩解症狀且無法顯著改變疾病進展,因此,尋找新的治療方法成為研究焦點。阿茲海默症的病理機制多樣化且複雜,其中腦部能量缺乏是顯著的病理特徵。J4是平衡型核苷轉運蛋白(ENT1)抑制劑,能透過改善腦部能量平衡來治療神經細胞能量缺損。臨床前實驗證實J4在不同阿茲海默症小鼠模型皆具有優異療效,能改善疾病小鼠認知功能障礙和空間記憶受損,減少異常聚集的Aβ和tau蛋白,減輕氧化壓力和神經發炎,並同時提高粒線體和葡萄糖代謝活性。對照實驗也證實J4比傳統臨床用藥donepezil和memantine之療效更為優越。研究團隊已完成J4臨床前藥動、藥效和安全性分析,正準備美國食品藥物管理局新藥臨床試驗(IND)許可所需的資料。J4作為具有新穎機制的口服小分子藥物,使用便利且成本經濟,良好療效及安全性使其具有極大發展潛力。J4已在多個國家取得專利,未來預計以新創團隊出場,與國內外藥廠及機構合作,透過募集資源或專利授權以加速臨床試驗。J4的研發將使阿茲海默症患者擁有更多治療選擇,有助於改善病情與生活品質,並減輕家人長期照護壓力和醫療支出。
Alzheimer's Disease (AD) is the most common neurodegenerative disorder in aging societies, affecting millions of people worldwide. As the population ages, the number of patients continues to rise, presenting an escalating healthcare challenge. Current clinical medications offer only symptomatic relief with limited efficacy and do not significantly halt the progression of the disease. Consequently, identifying new therapeutic strategies is a primary focus of research. The pathogenesis of AD is intricate, with brain energy deficit being a key contributor to AD onset. J4, an equilibrative nucleoside transporter 1 (ENT1) inhibitor, presents a novel mechanism to rectify energy imbalances in the brain and address neuronal energy deficits. Preclinical studies have shown remarkable therapeutic effects of J4 in various AD mouse models. These effects include improvement in cognitive impairments and spatial memory deficits, reduction in the abnormal accumulation of Aβ and tau proteins, alleviation of oxidative stress and neuroinflammation, and enhancement of mitochondrial and glucose metabolism activity. J4 has demonstrated superior efficacy compared to traditional treatments like donepezil and memantine. The research team has completed the preclinical pharmacokinetics, pharmacodynamics, efficacy, and safety analysis of J4 and is now compiling the necessary documentation for the Investigational New Drug (IND) application to the U.S. Food and Drug Administration (FDA) for the commencement of clinical trials. As an orally administered small molecule drug with a novel mechanism, J4 holds substantial development potential due to its convenience, cost-effectiveness, and promising efficacy and safety. With patents secured in multiple countries, J4 is poised for development by a new startup team. This team will collaborate with domestic and international pharmaceutical companies and institutions to accelerate clinical trials, either through resource mobilization or patent licensing. The advancement of J4 will provide AD patients with a superior treatment option, enhancing their quality of life and alleviating the financial and caregiving burdens faced by their families.
-
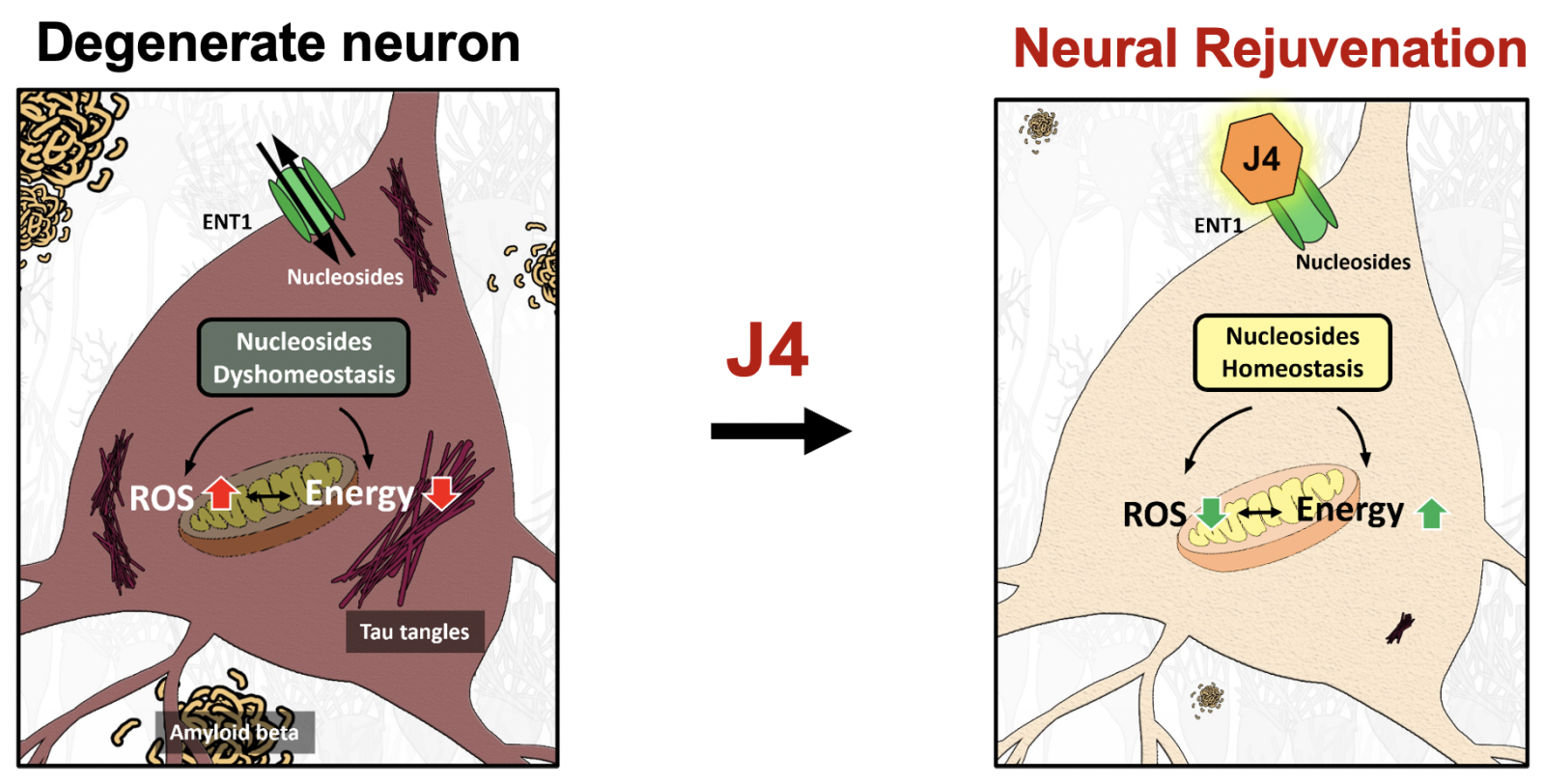
ENT1 is an important membrane transporter responsible for the uptake of nucleosides (e.g., adenosine, inosine, nicotinamide riboside) in the brain, and plays critical roles in modulating neuronal activity and energy metabolism. J4 is a first-in-class compound, serving as an ENT1 inhibitor, that helps to address the problem of low energy supply in Alzheimer’s disease.
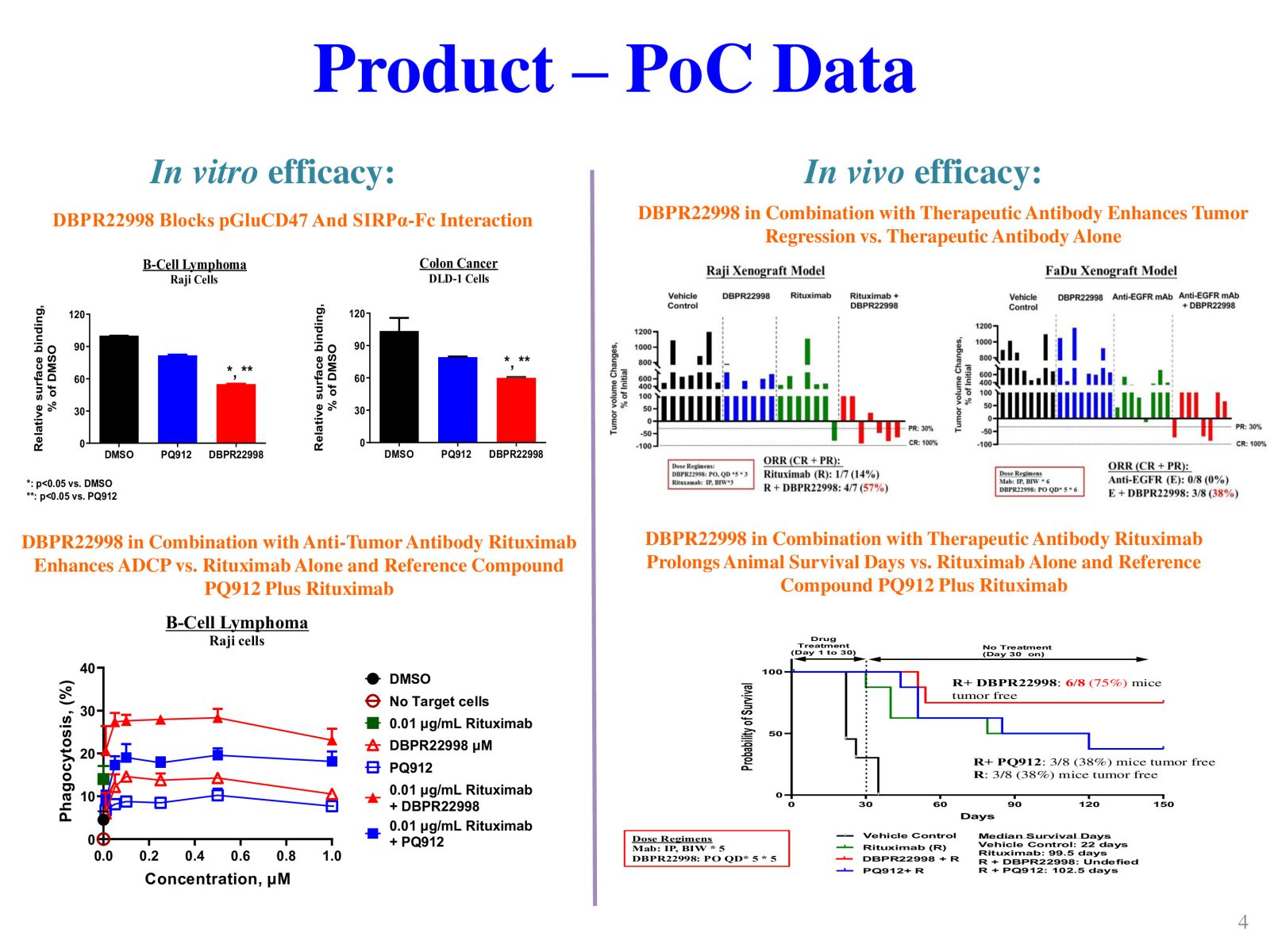
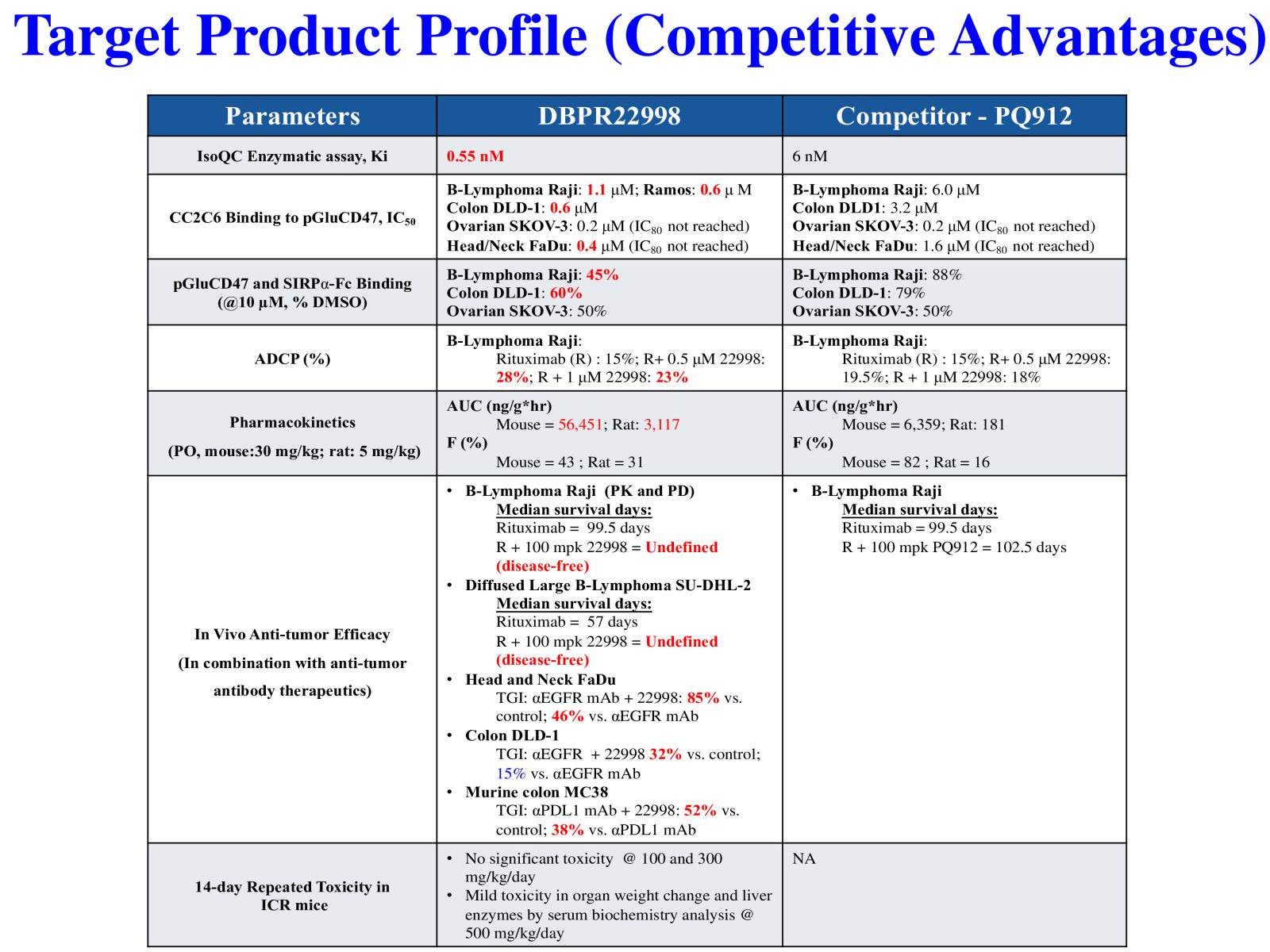
4DBPR22998:以 CD47-SIRPα “別吃我“ 信號為標的之新型廣效癌症免疫治療藥物國家衛生研究院生技與藥物研究所 National Health Research Institutes, Taiwan顏婉菁研究員發明人:陳志豪, 顏婉菁, 葉燈光, 陳炯東, 王惠鈞, 黃開發領域:新穎藥物適應症:Cancer研發階段:候選藥物/醫材雛型試製造 Pilot production of candidate drug/prototype摘要:DBPR22998 為癌症免疫治療領域首創之新穎、口服用的小分子候選藥物,靶向CD47蛋白合成後修飾過程,可有效抑制isoQC的酵素活性,進而減少腫瘤細胞表面上CD47與SIRPα的結合以及“別吃我” 免疫檢查點訊息傳導。與單株抗體標靶藥物或免疫檢查點抗體合併使用可促進抗體依賴性細胞吞噬以及提高腫瘤消除作用,進而達到治療癌症的效果。小分子isoQC 抑制劑不會引起貧血或血小板減少症,因此避免了CD47單株抗體治療潛在的副作用以及風險。DBPR22998 物質專利目前已獲美國、中華民國等八國之核准; 癌症適應症全球專利中華民國專利已核准,其他專利審查中。未來將與產業以技術移轉連結, 或與國內外大藥廠產學合作共同推動臨床試驗,以治療容易復發且耐藥性高之癌症病患。
DBPR22998 is a novel oral small molecule cancer immunotherapeutic drug candidate targeting the post-translational modification process of CD47 protein to inhibit CD47-SIRPα “ Do not eat me” immune checkpoint singal. DBPR22998 effectively inhibits the enzymatic activity of isoQC and interferes with the binding of tumor cell surface CD47 and SIRPα on the macrophage. DBPR22998 in combination with monoclonal antibody therapeutics or immune checkpoint inhibitors enhances antibody-dependent cellular phagocytosis and improves tumor elimination in murine tumor models. Of note, DBPR22998 does not cause anemia or thrombocytopenia and thus avoids the potential side effects and risks of CD47 monoclonal antibody therapeutics. The DBPR22998 substance patents have been approved by eight countries including the United States and the Republic of China; the PCT patents for cancer indication has been approved by the Republic of China and other patents are under the review. In the future, we will connect with the industry through technology transfer or in collaboration with pharmaceutical companies to jointly promote clinical trials to treat cancer patients who are prone to relapse and have high drug resistance.
-
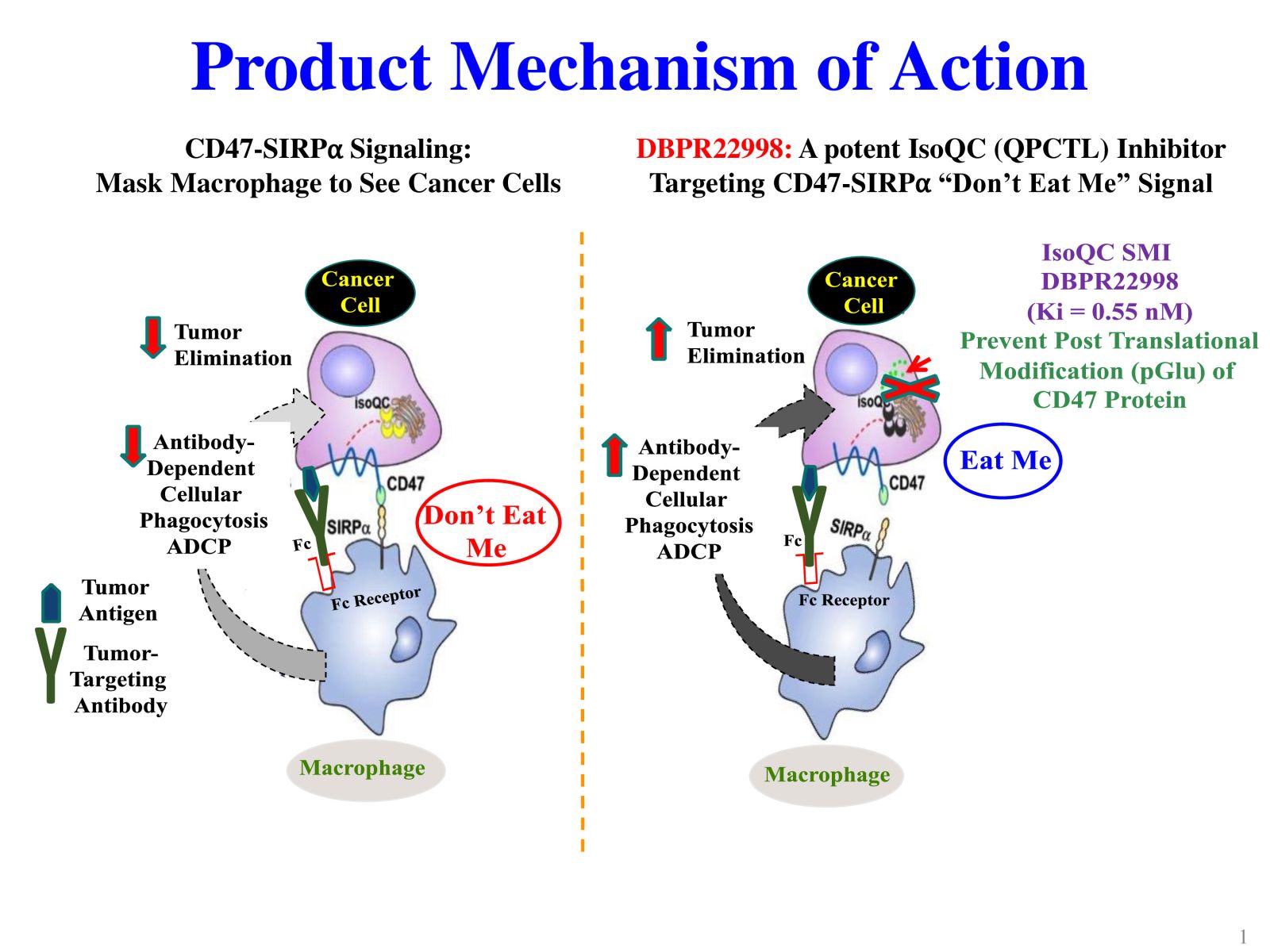



Slide 1: Product Mechanism of Action
Slide 2. Advantages of Targeting IsoQC Over Targeting CD47
Slide 3. Product Summary
Slide 4. Product- PoC data
Slide 5. Target Product Profile (Competitive Advantages)
Slide 6. Product Development and Patent Portfolio
5預防角膜內皮細胞流失導致的角膜失明:市場首見小分子藥物國立臺灣大學醫學工程學系林頌然教授兼系主任發明人:林頌然領域:新穎藥物適應症:Corneal endothelial cell loss after transcorneal/intraocular surgeries研發階段:先導藥物最佳化 Lead drug optimization摘要:目前,全世界約有1,270萬名患者等待角膜移植,其中台灣約有1,000名患者在等待名單上。 角膜內皮功能障礙佔角膜移植病例的50%以上。 對於角膜內皮失代償的創新治療存在著巨大的未滿足的需求。 生理上,由於增殖能力有限,角膜內皮細胞每年損失 0.6%。 然而,經角膜/眼內手術,例如白內障和青光眼手術,會加速角膜內皮細胞損失率,範圍為 10% 至 20%。 最近,我們鑑定了一種小分子藥物,化合物A,其分子量小於400 kDa。 在小鼠模型中,前房注射 1 ng 化合物 A 已顯示出三種明顯的益處,包括促進傷口癒合、保持角膜透明度以及預防損傷導致的進行性角膜內皮細胞損失。 因此,我們進一步產品開發的策略是在閉合手術切口傷口之前將化合物 A 遞送至房水中。 重要的是,這種方法不會造成額外的傷害,並最大限度地減少眼科醫生的工作量。 我們的研究結果提出了預防眼內手術引起的角膜內皮細胞損失的創新轉譯療法。
Currently, there are approximately 12.7 million patients globally awaiting corneal transplantation, with about 1,000 patients in Taiwan on the waiting list. Corneal endothelial dysfunction accounts for more than 50% of cases of corneal transplantation. A significant unmet need exists for innovative treatments for corneal endothelium decompensation. Physiologically, corneal endothelial cell loss at 0.6% yearly due to limited proliferation capacity. However, transcorneal/intraocular surgeries, such as cataract and glaucoma surgeries, accelerate corneal endothelial cell loss rates, ranging from 10% to 20%. Recently, we have identified a small molecular drug, compound A, with a molecular weight of less than 400 kDa. In a mouse model, the intracameral injection of 1ng compound A has demonstrated three distinct benefits, including promoting wound healing, maintaining corneal transparency, and halting progressive corneal endothelial cell loss from injury. Accordingly, our strategy for further product development is to deliver compound A into the aqueous humor before closing the surgical incisional wound. Importantly, this approach imposes no additional injuries and minimizes the workload for ophthalmologists. Our findings present innovative translational treatments for preventing corneal endothelial cell loss from intraocular surgeries.
-
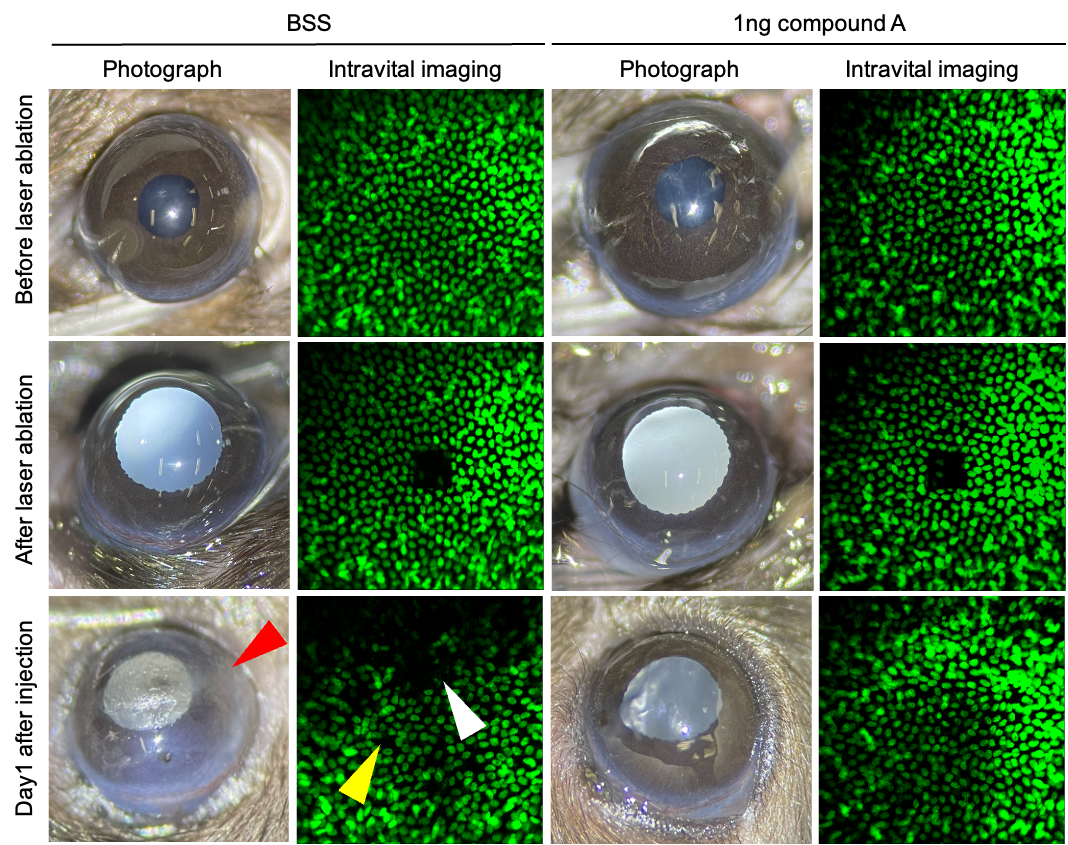
Intracameral injection of compound A promotes wound healing (white arrowhead), maintains corneal transparency (red arrowhead) and prevents corneal endothelial cell loss (yellow arrowhead).
6Develop a Novel Monoclonal Antibody for the therapy of rheumatoid arthritis中國醫藥大學醫學系蔡嘉哲教授發明人:蔡嘉哲領域:新穎藥物適應症:治療對疾病緩解型抗風濕性藥物及生物製劑無適當療效之類風濕性關節炎患者研發階段:動物驗證 In vivo validation摘要:現有治療類風濕關節炎的生物製劑包含anti-TNF已大幅改善類風濕關節炎的治療效果。然而,仍有20-30%類風濕關節炎患者對第一線生物製劑治療反應不佳。目前藥物設計皆為修飾或抑制免疫反應,因此有必要發展不同標的模式的生物製劑。我們研發一新穎單株抗體用於治療類風濕關節炎,以牙周病細菌的一段胜肽作為抗原,利用老鼠產生單株抗體,再以膠原蛋白誘導類風濕關節炎動物模型(CIA)進行治療用單株抗體效力測試。我們鑑定新開發的單株抗體與免疫胜肽成專一性反應,在動物模型實驗顯示,單株抗體治療CIA動物具劑量依賴性。治療後改善了關節指數及踝圍腫脹,關節間隙及軟骨破壞,並減緩發炎反應及滑膜增生。總結,此新穎性單株抗體治療在CIA動物模型應用優於現行用藥Enbrel。
Existing biologics for the treatment of rheumatoid arthritis, including anti-TNF agents, have significantly improved the therapeutic outcomes. However, approximately 20-30% of rheumatoid arthritis patients exhibit poor responses to first-line biologic therapies. Currently, drug designs primarily focus on modifying or suppressing the immune response, necessitating the development of biologics targeting different pathways. We have developed a novel monoclonal antibody for the treatment of rheumatoid arthritis, utilizing a peptide derived from periodontal bacteria as the antigen. Monoclonal antibodies were generated in mice and subsequently evaluated for their therapeutic efficacy in a collagen-induced arthritis (CIA) animal model of rheumatoid arthritis. We identified the newly developed monoclonal antibody to exhibit specific reactivity with the immunopeptide. In animal model experiments, the monoclonal antibody demonstrated dose-dependent effects in treating CIA animals. Following treatment, improvements were observed in joint scores, ankle swelling, joint space, cartilage damage, and a reduction in inflammatory responses and synovial hyperplasia. In summary, this novel monoclonal antibody therapy outperforms the current medication Enbrel in the CIA animal model application, showing promising potential for the treatment of rheumatoid arthritis.
-
.jpg)
1. 我們從研究牙周病與類風濕性關節炎發現一段胜肽為診斷類風濕性關節炎的新穎抗原。2. 利用此胜肽產生一單株抗體,其為IgG1 kappa抗體,與免疫胜肽BR1(類風濕性關節炎抗原決定位)成專一性反應,並且和FCγRIIb及 FCγR III具結合能力,尤其FCγRIIb具有高度結合力。3. 此單株抗體在類風濕性關節炎動物模型CIA有良好的治療效果。4. 此單株抗體將進入臨床試驗。
7治療腸躁症腹痛的首創型新藥國立臺灣大學創新藥物研究中心Center for Innovative Therapeutics Discovery, National Taiwan University忻凌偉副教授兼主任發明人:忻凌偉/Ling-Wei Hsin領域:新穎藥物適應症:腸躁症腹痛/abdominal pain in irritable bowel syndrome研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:腸躁症是一種常見的功能性腸胃障礙症候群,其特徵是反覆發作的腹痛伴隨著排便習慣改變,目前還找不出確切病因。嚴重腹痛常是腸躁症病人求診的主因,但目前藥物多是針對腹瀉或便秘症狀緩解,尚無令人滿意的腸躁症腹痛治療藥物,仍為高度未獲滿足的醫療需求。DC105是經由全新的標靶與藥理機轉的首創型新成分新藥,具有強效的內臟止痛效果與良好的安全性,已取得美國、中華民國、日本及韓國專利。若DC105能夠順利發展為治療腸躁症腹痛的首創型新藥,2040年之前,每年銷售額預期最少可達10億美金。將採用成立新創公司募資並在適當時機進行技術轉移的策略,經由三個階段性目標:通過研究性新藥申請、完成臨床一期試驗及完成臨床二期試驗,達成DC105的商品化。
Irritable bowel syndrome (IBS) is a chronic functional gastrointestinal disorder characterized by recurrent abdominal pain and changes in bowel habits with unknown etiology. The severe abdominal pain is the most likely complaint to result in medical consultation. Currently, the available drugs in clinical for IBS patients are effective for the altered bowel movement, but none of them is satisfactory for the treatment of intestinal pain in IBS. The abdominal pain in IBS is a highly unmet need. DC105 is a first-in-class, new chemical entity drug targeting a new biological target and pharmacological mechanism. It has a strong visceral analgesic effect and high safety. The United States, Taiwan, Japan, and Korea Patents for DC105 have been granted. If DC105 can be successfully developed into a first-in-class new drug for the treatment of abdominal pain in IBS, the annual sales are expected to be at least 1 billion USD before 2040. Establishment of a new startup company to raise funds and conducting technology transfer at the appropriate time will be adopted as our strategies to achieve the commercialization of DC105, through three phased goals: approval of investigational new drug (IND) application, completion of Phase I clinical trials and completion of Phase II clinical trials.
-
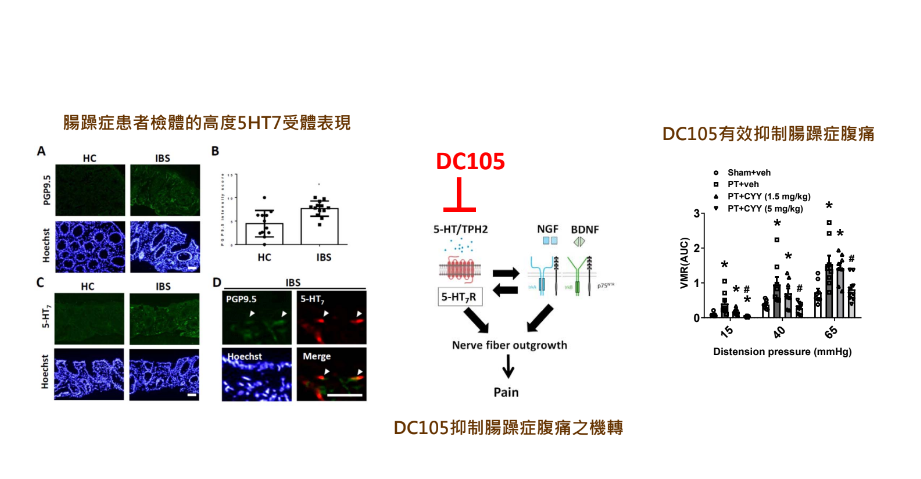
DC105的生物標靶、作用機轉及止痛效果。左:腸躁症患者的腸黏膜檢體具有昇高的5HT7受體表現量;中:DC105抑制腸躁症腹痛的藥理作用機轉;右:DC105在腸躁症動物模式中,有效抑制腸躁症的腹痛。
8開發新標靶神經降壓素受體(NTSR1)蛋白之抗體藥物複合體(ADC)用於頭頸癌治療財團法人生物技術開發中心/生物製藥研究所蔡士昌所長發明人:游成州領域:新穎藥物適應症:頭頸癌研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:過去,頭頸癌的治療主要使用類鉑化合物(如Cisplatin)和5-FU等化療藥物,而整體反應率仍然僅為30-40%,且復發和轉移的風險仍然高,說明頭頸癌治療仍有改進的空間,需要更有效的方法來提高患者的預後。根據文獻報告,NTSR1蛋白在多種惡性腫瘤的發展中扮演重要角色,在頭頸癌患者中,超過50%的患者高度表現NTSR1蛋白,可能增加癌症發展和轉移的風險,並與不良預後相關,因此,NTSR1被視為頭頸癌治療的重要標靶,可用於新型治療藥物的研究和開發。為此,國衛院和生技中心聯合開發了新型αNTSR1-ADC藥物,該藥物已在細胞和動物模型中展現出良好的藥代動力學和卓越的療效,同時在生產細胞株開發方面也具有高產能的優勢。這些結果確認了新型αNTSR1-ADC藥物的創新性、進步性和競爭力。
Traditional chemotherapy drugs, such as cisplatin and 5-fluorouracil, have been used to treat head and neck cancer, but the overall response rate is only 30-40%, and recurrence and metastasis rates remain high. This indicates that there is still a need for more effective treatments for head and neck cancer. According to literature reports, the NTSR1 protein plays an important role in the development of various malignant tumors, including head and neck cancer. Over 50% of head and neck cancer patients show high levels of NTSR1 protein expression, which may increase the risk of cancer development and metastasis and is associated with poor patient prognosis. NTSR1 is considered an important target for head and neck cancer treatment and may be used as a direction for the development of new treatments. To this end, the NHR1 and DCB have collaborated to develop a new αNTSR1-ADC drug for head and neck cancer treatment. The drug uses a combination of αNTSR1 antibody and trimannosyl ADC platform and has shown excellent PK and therapeutic efficacy in cancer cells and animal models, as well as high production yield in CHO cell line development. These results confirm that the new αNTSR1-ADC drug is novel, progressive, and competitive.
-
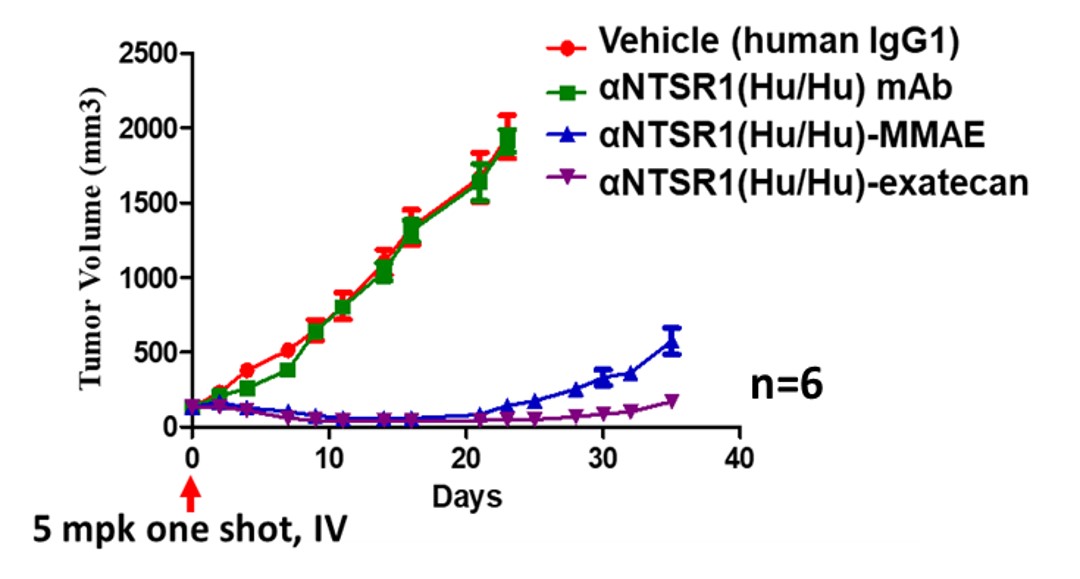
Targeting NTSR1 in Squamous Cell Carcinoma of the Head and Neck: Preclinical Proof-of-Concept with the Investigational Anti-NTSR1 Antibody-drug Conjugate display potent antitumor activities.
9靶向抗原國家衛生研究院感染症與疫苗研究所陳信偉研究員發明人:陳信偉、冷治湘、劉士任領域:新穎藥物, 劑型開發適應症:癌症治療、疫苗開發研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:疫苗開發面臨一項重大挑戰,即在體內有效地將抗原遞送到樹突狀細胞(DCs),實現交叉呈遞並誘導記憶免疫反應。Fcγ受體(FcγRs)存在於多種細胞類型中,包括DCs,因此通過FcγRs將抗原遞送到DCs,成為疫苗開發中一種具吸引力的策略。本研究利用FLIPr(金黃葡萄球菌所分泌一種與FcγRs結合的蛋白),用於將抗原傳遞給DCs。我們的研究結果顯示,FLIPr能夠有效將抗原傳遞給CD8+ DCs,誘導出強大的免疫反應,無需額外的輔助劑。當抗原與FLIPr融合時,能夠有效將抗原經由MHC II和MHC I呈遞,活化抗原專一性之CD4與CD8 T細胞,並誘導記憶T細胞反應的生成。總之,利用FLIPr作為抗原傳遞載體對於發展癌症免疫療法以及對抗傳染病的疫苗具有巨大潛力。
Vaccine development faces a significant challenge in efficiently targeting antigens to dendritic cells (DCs) within the body, enabling cross-presentation and the generation of memory immune responses. Fcγ receptors (FcγRs) are present on various cell types, including DCs, making the targeting of antigens to DCs through FcγRs an appealing strategy in vaccine development. This technology utilizes the formyl peptide receptor-like 1 inhibitory protein (FLIPr), a protein secreted by Staphylococcus aureus that binds to FcγRs, as a means to deliver antigens to DCs. We demonstrate that FLIPr effectively delivers antigens to CD8+ DCs, inducing robust immune responses without the need for additional adjuvants. When antigens are fused with FLIPr, efficient antigen presentation occurs on both MHC class II and class I, leading to the generation of memory T cell responses. In summary, employing FLIPr as an antigen delivery vector holds significant promise for the development of cancer immunotherapies and vaccines against infectious diseases.
-
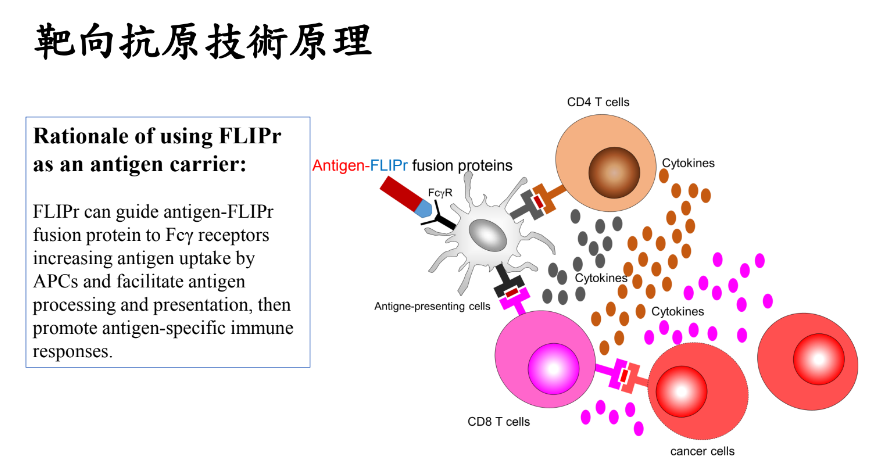
靶向抗原技術原理:利用金黃葡萄球菌所分泌之FLIPr與FcγR結合之特性,將抗原有效傳遞到樹突細胞,進而增強免疫反應。
10新穎功能性益生菌開發:利用腸道菌改善高雄性素血症中央研究院生物多樣性研究中心江殷儒副主任/研究員發明人:江殷儒、陳美州、蕭尊先、周佳宏領域:新穎藥物, 益生菌保健食品適應症:高雄性素血症相關疾病,hyperandrogenism-related diseases研發階段:動物驗證 In vivo validation摘要:高雄性素血症乃指血液中存在過量的雄性素,並和雄性禿、攝護腺肥大及攝護腺癌,及多囊性卵巢症候群有關。團隊發現,人體及動物腸道中的陶爾氏菌(Thauera),具有代謝雄性素的特殊能力;我們解析其相關代謝基因以了解此生化反應機制。透過小鼠管餵試驗,我們觀察腸道菌叢變化及檢測血清生化數值。實驗中,該菌對小鼠未造成不良反應且能有效降低血清中雄性素濃度。同時透過比較基因體學及培養體學的方式,我們也分離出能代謝雄性素的乳酸菌株。初步的小鼠實驗也確認這些菌株能改善雙氫睪酮造成的毛髮生長遲緩。其中部分菌株屬於衛服部食藥署所規範之可為食品的正面表列菌種。未來希望推出功能性腸道菌,並提供針對人體高雄性素血症相關病症之輔助治療策略。
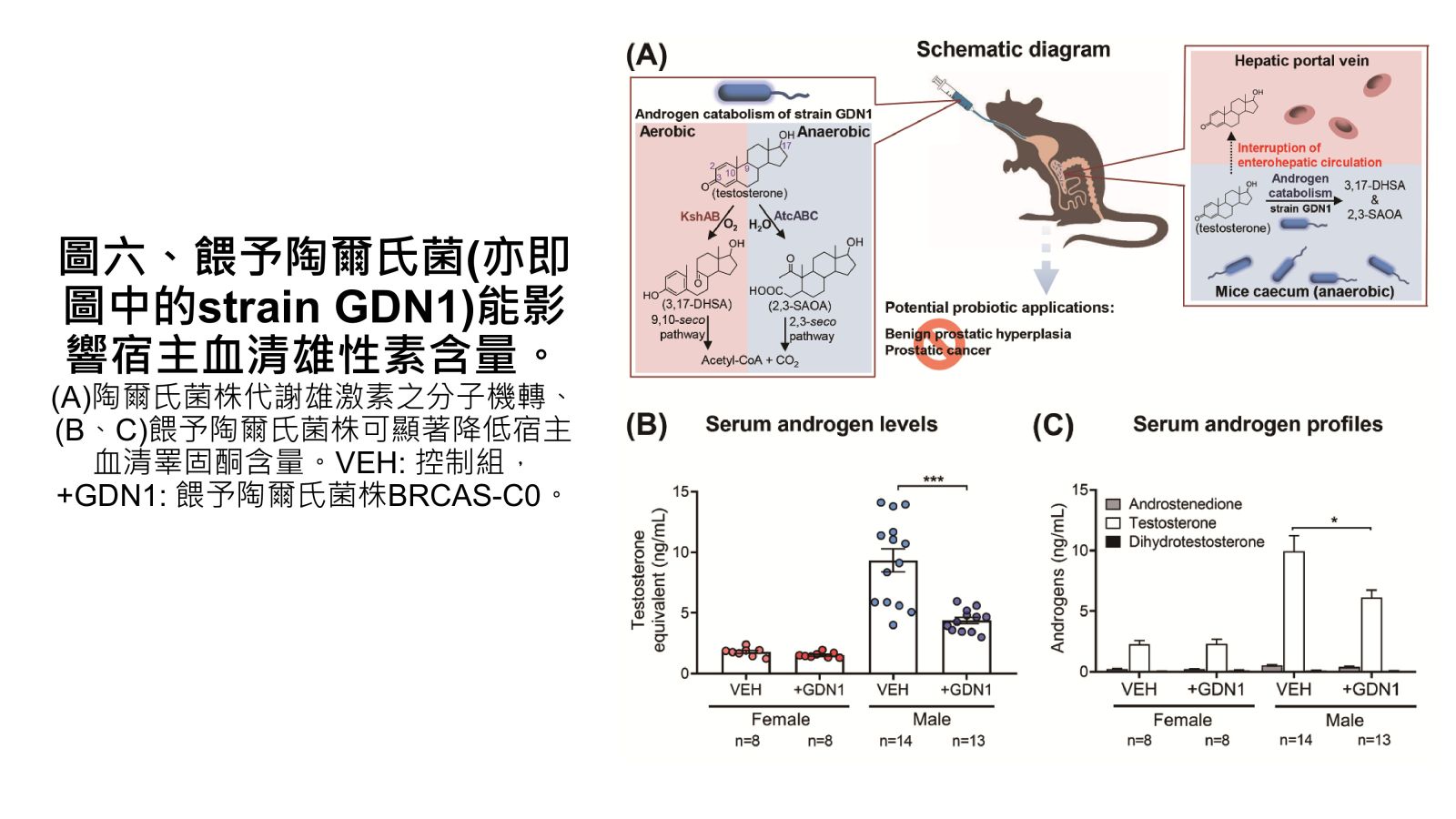
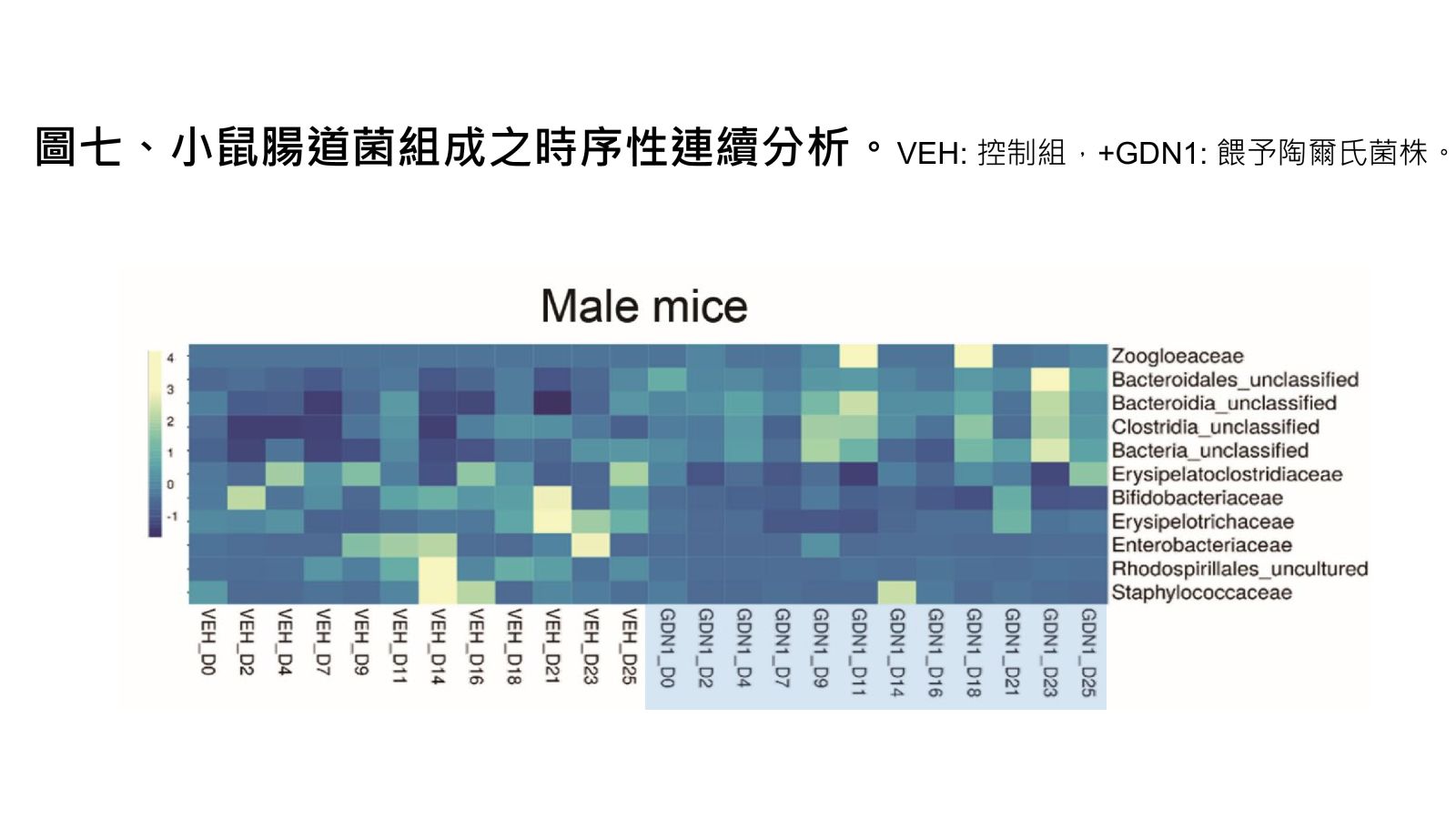
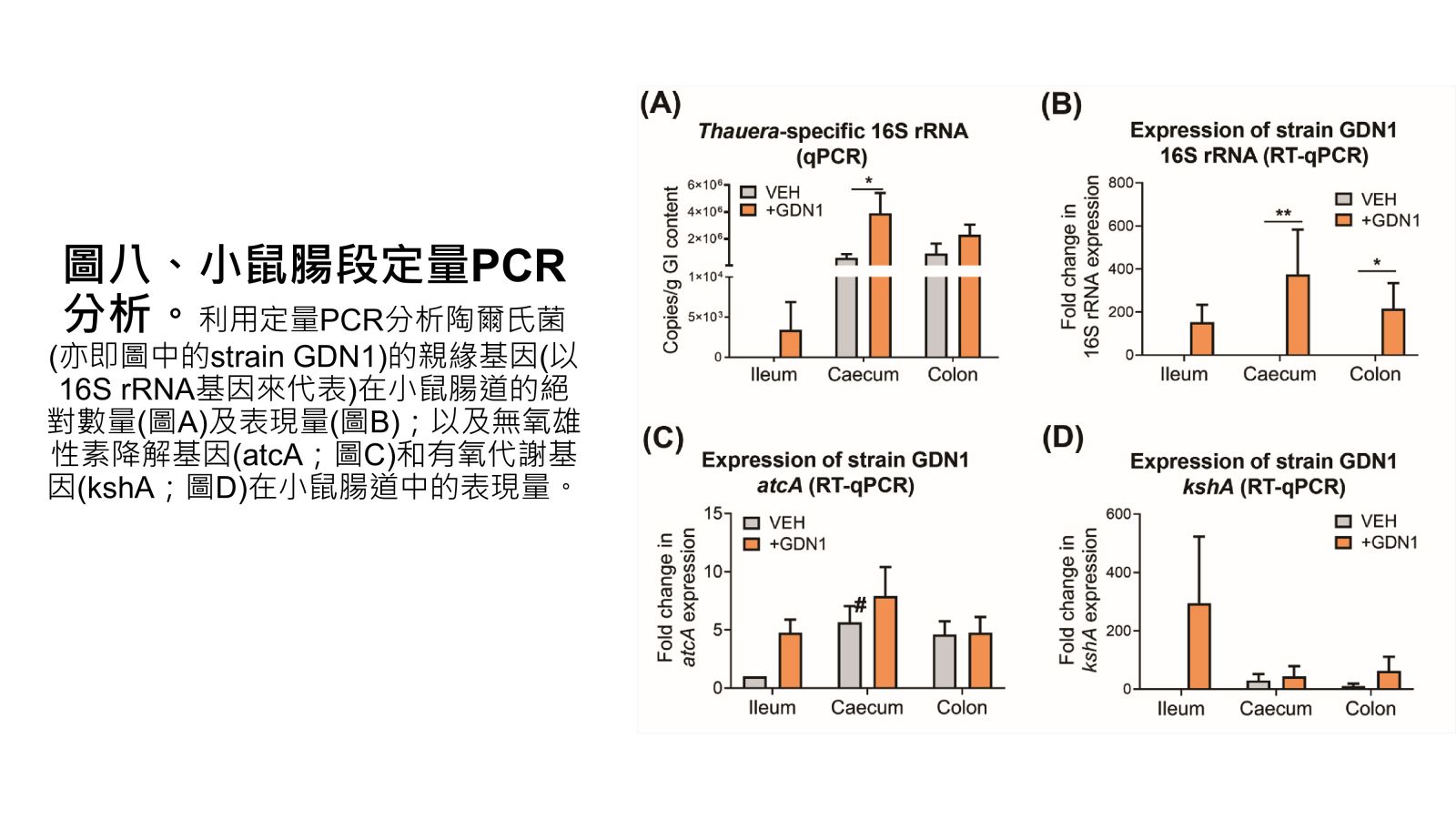
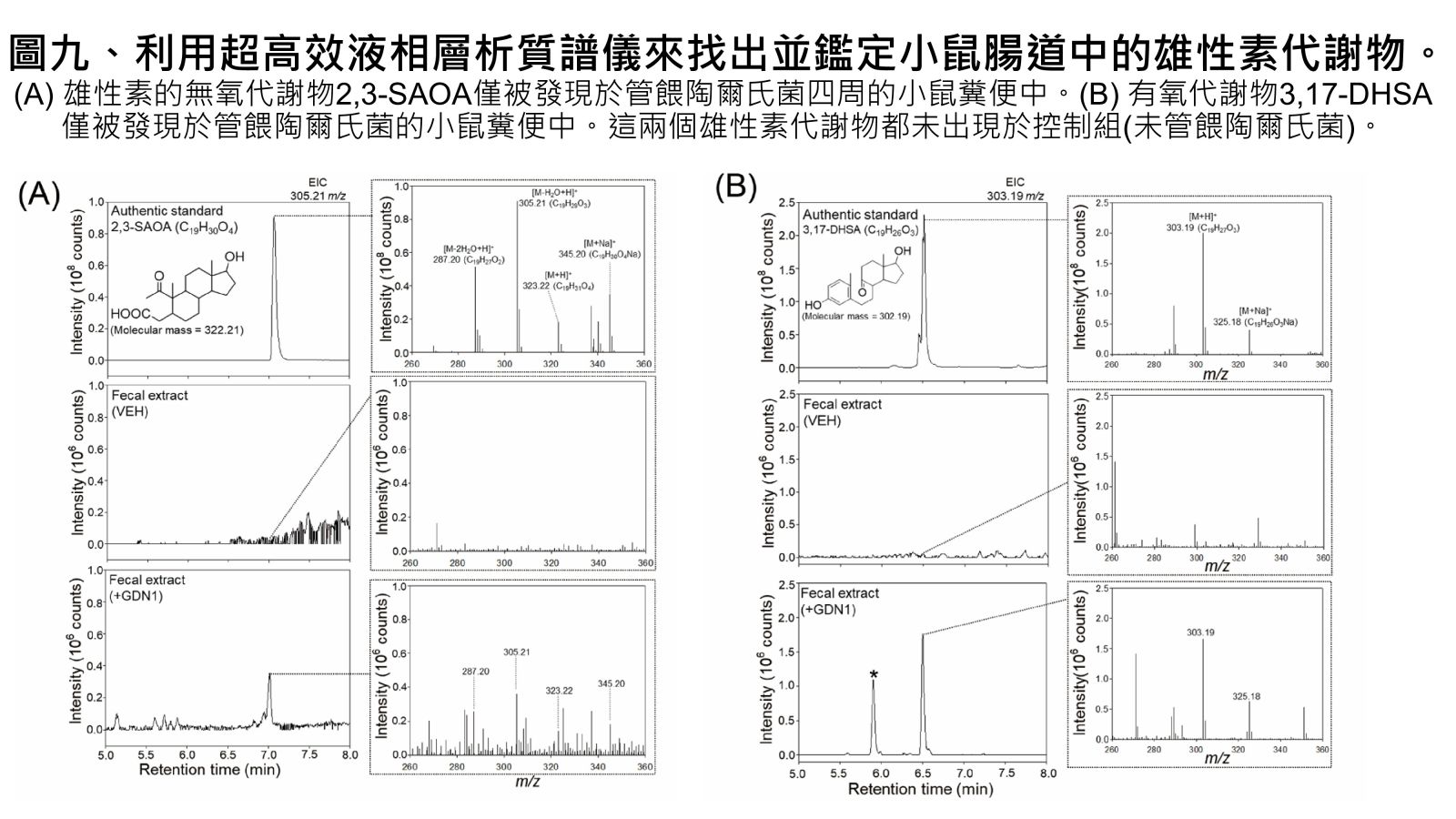
Vertebrates, including humans, can synthesize a variety of sex hormones; however, animals cannot completely degrade these steroids. Sex hormones are recycled between the liver and the gut through enterohepatic circulation; the reabsorption of these steroids occurs mainly in human small intestine as well as rodent caecum. Gut microbes may modify and degrade gut steroids and thus regulate host sex hormone levels and profiles. Abnormally high circulating androgen levels have been considered a causative factor for benign prostatic hypertrophy and prostate cancer in men. Recent animal studies on gut microbiome suggested that gut bacteria are involved in sex steroid metabolism; however, the underlying mechanisms and bacterial taxa remain elusive. Denitrifying betaproteobacteria Thauera spp. are metabolically versatile and often distributed in the animal gut. Thauera sp. strain GDN1 is an unusual betaproteobacterium capable of catabolizing androgen under both aerobic and anaerobic conditions. We administered C57BL/6 mice (aged 7 weeks) with strain GDN1 through oral gavage. The strain GDN1 administration caused a minor increase in the relative abundance of Thauera (≤ 0.1%); however, it has profound effects on the host physiology and gut bacterial community. The results of our ELISA assay and metabolite profile analysis indicated an approximately 50% reduction in serum androgen levels in the strain GDN1-administered male mice. Moreover, androgenic ring-cleaved metabolites were detected in the fecal extracts of the strain GDN1-administered mice. Furthermore, our RT–qPCR results revealed the expression of the androgen catabolism genes in the gut of the strain GDN1-administered mice. We found that the administered strain GDN1 regulated mouse serum androgen levels, possibly because it blocked androgen recycling through enterohepatic circulation. This study discovered that sex steroids serve as a carbon source of gut bacteria; moreover, host circulating androgen levels may be regulated by androgen-metabolizing gut bacteria. Similar functional genes and corresponding androgen-transforming enzymes have been identified in our lactic acid bacteria. Among them, some probiotics can be used directly as food additives in Taiwan. We confirmed their androgen-metabolizing capability through physiological tests. Moreover, administration of these bacterial strains (BRCAS-C1~BRCAS-C3) could apparently alleviate the hair loss caused by dihydrotestosterone, the most potent androgen. Our data thus indicate the possible applicability of androgen-metabolizing gut bacteria as potent probiotics in alternative therapy of hyperandrogenism.
-
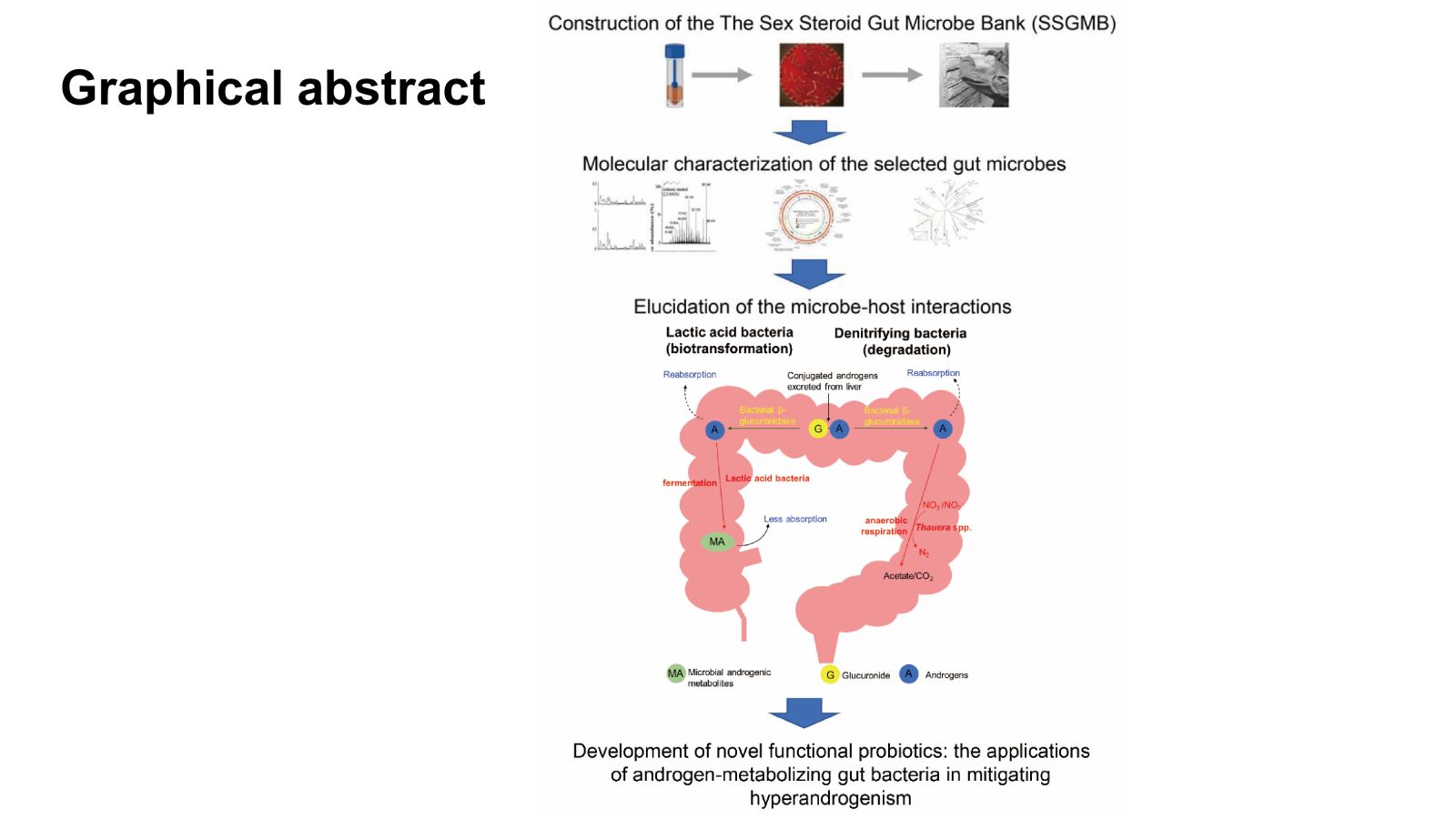
Graphical abstract
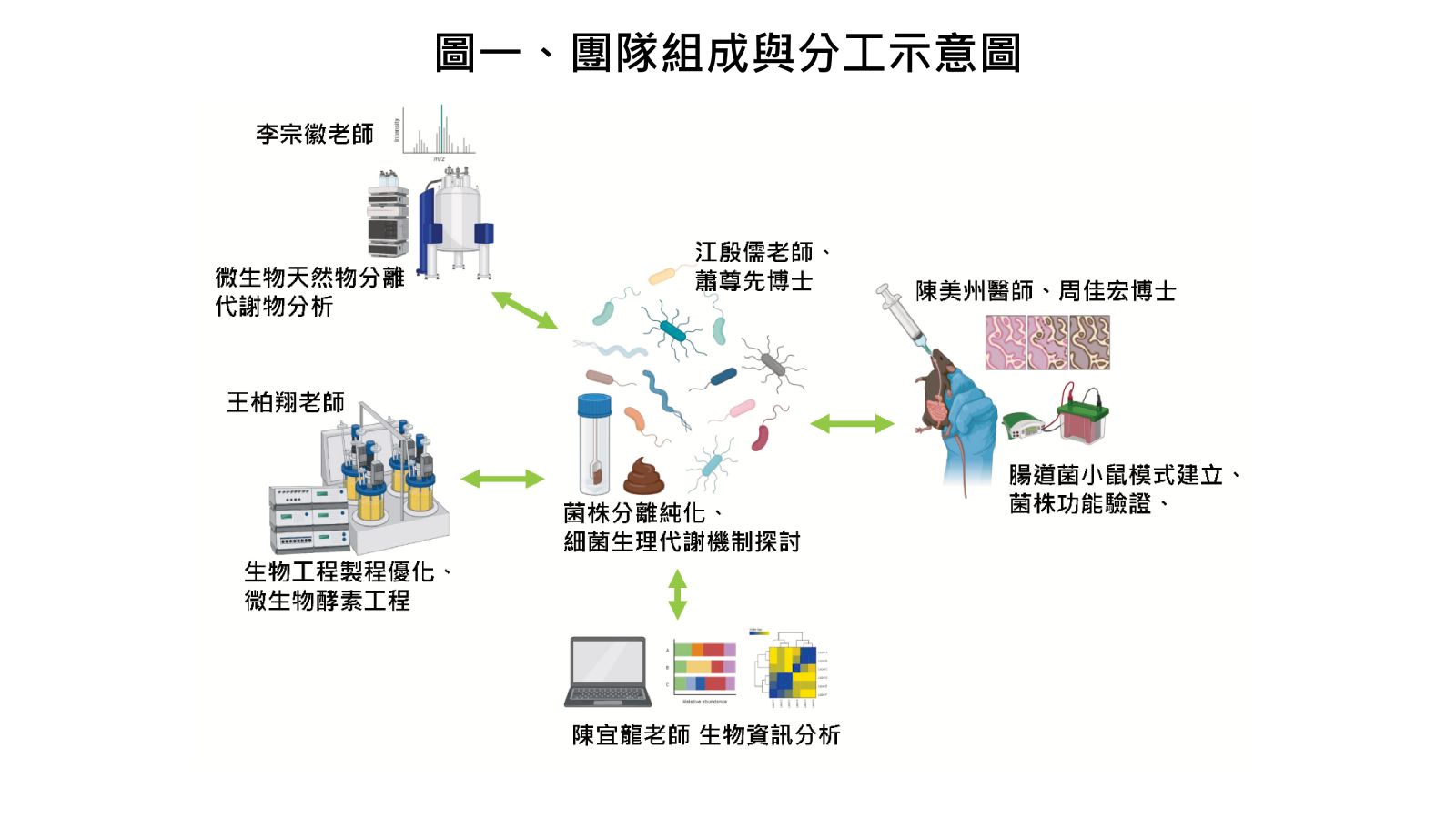
圖一、團隊組成與分工示意圖
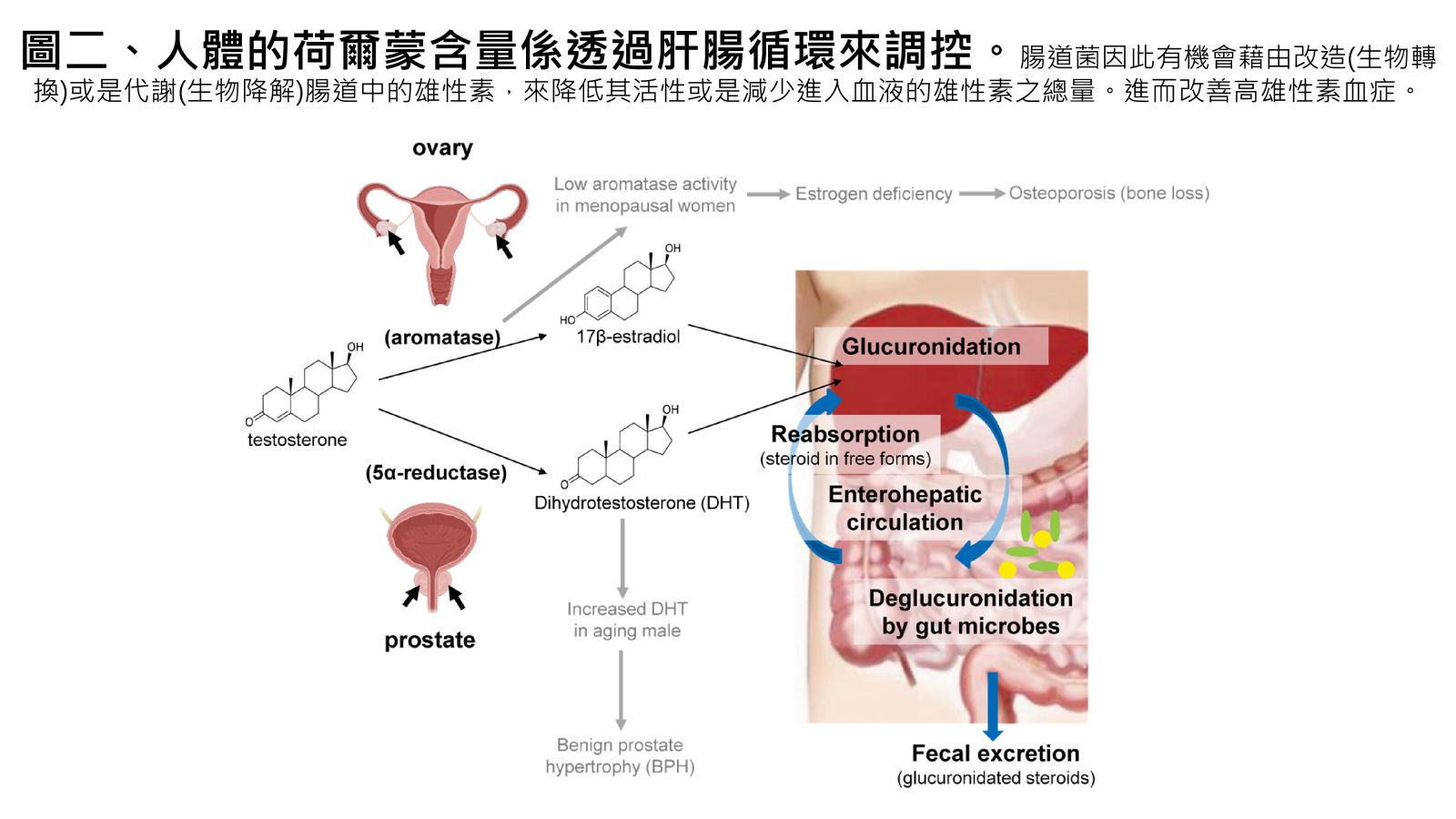
圖二、人體的荷爾蒙含量係透過肝腸循環來調控。
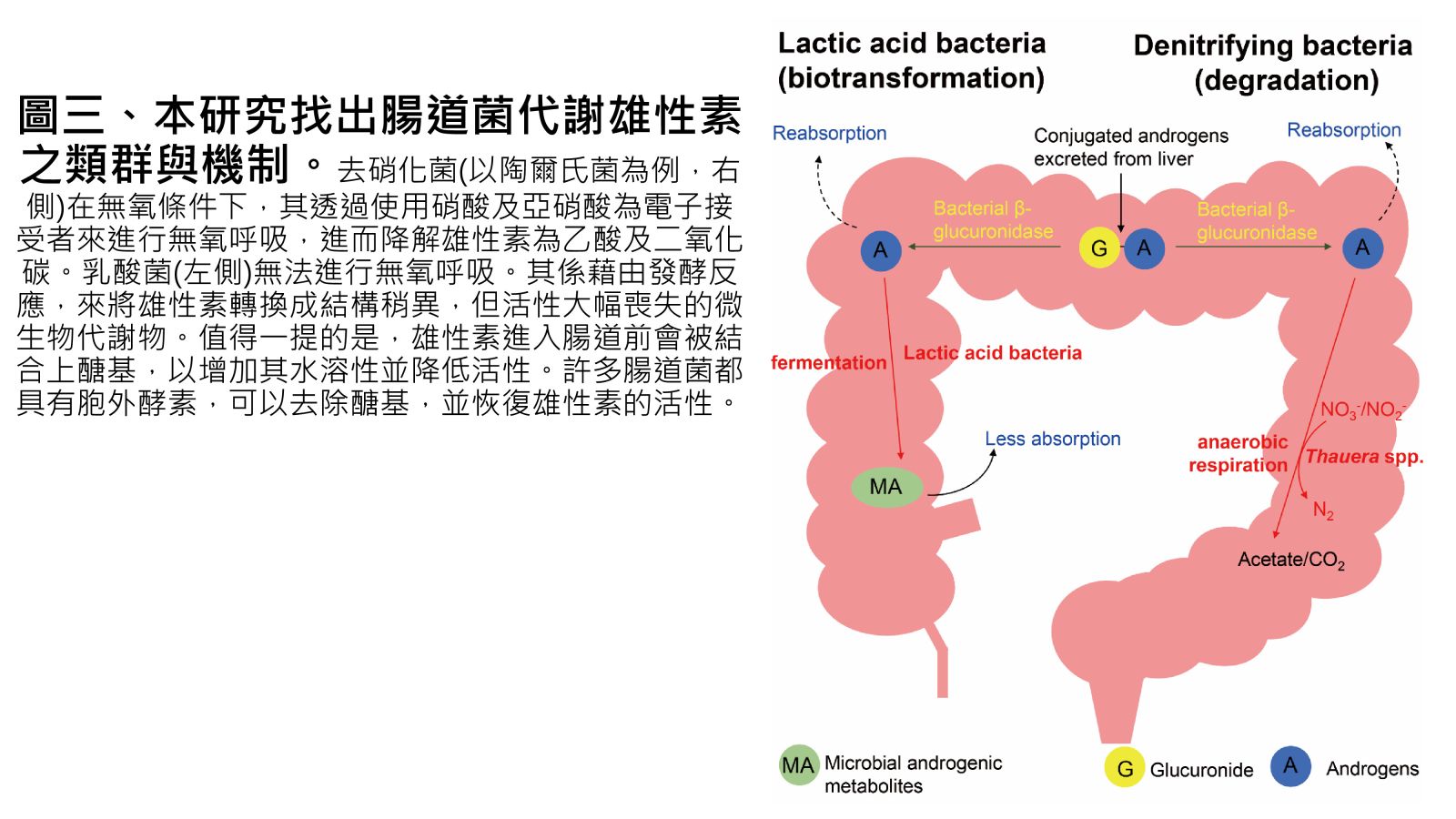
圖三、本研究找出腸道菌代謝雄性素之類群與機制。
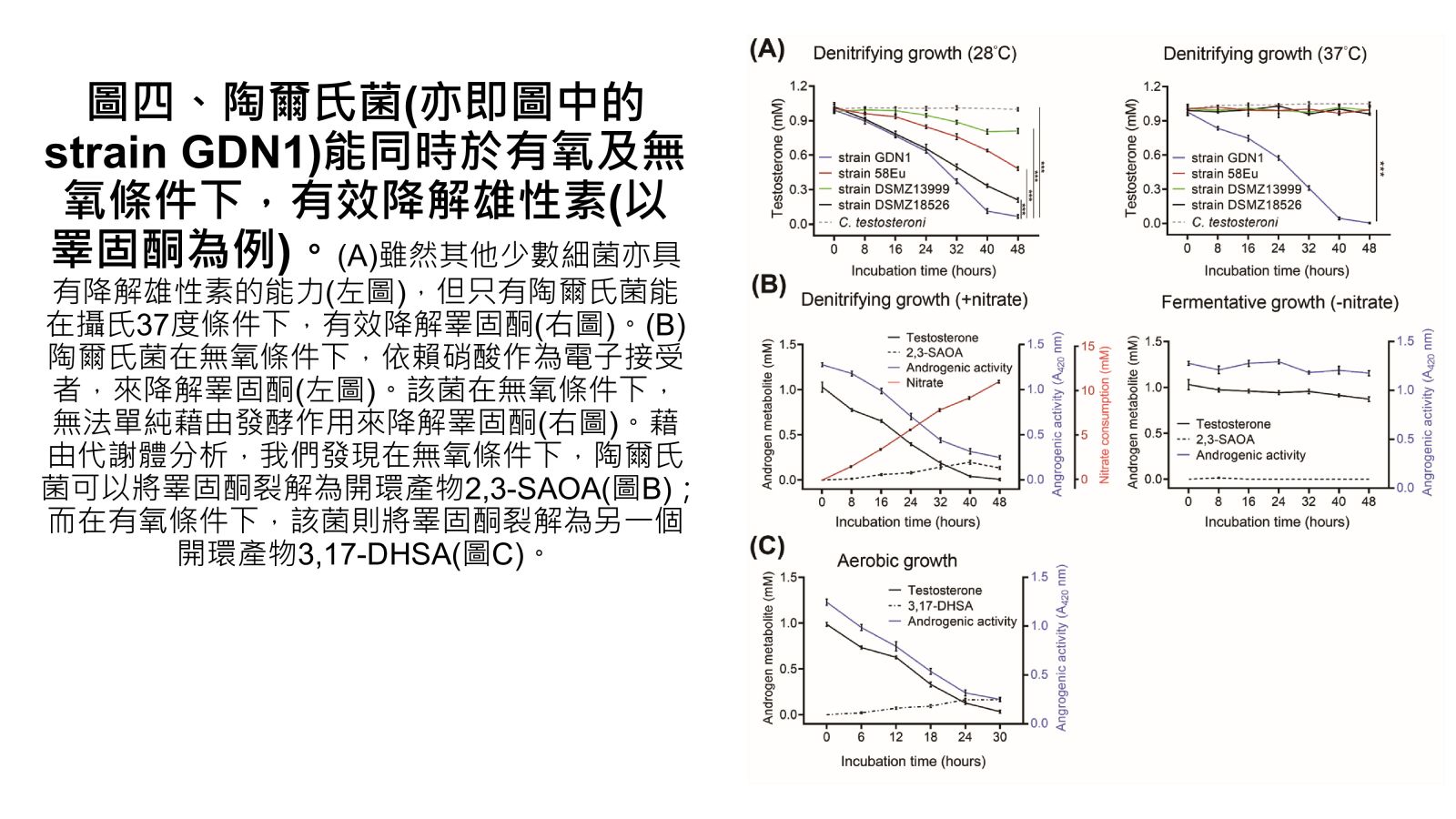
圖四、陶爾氏菌(亦即圖中的strain GDN1)能同時於有氧及無氧條件下,有效降解雄性素(以睪固酮為例)。
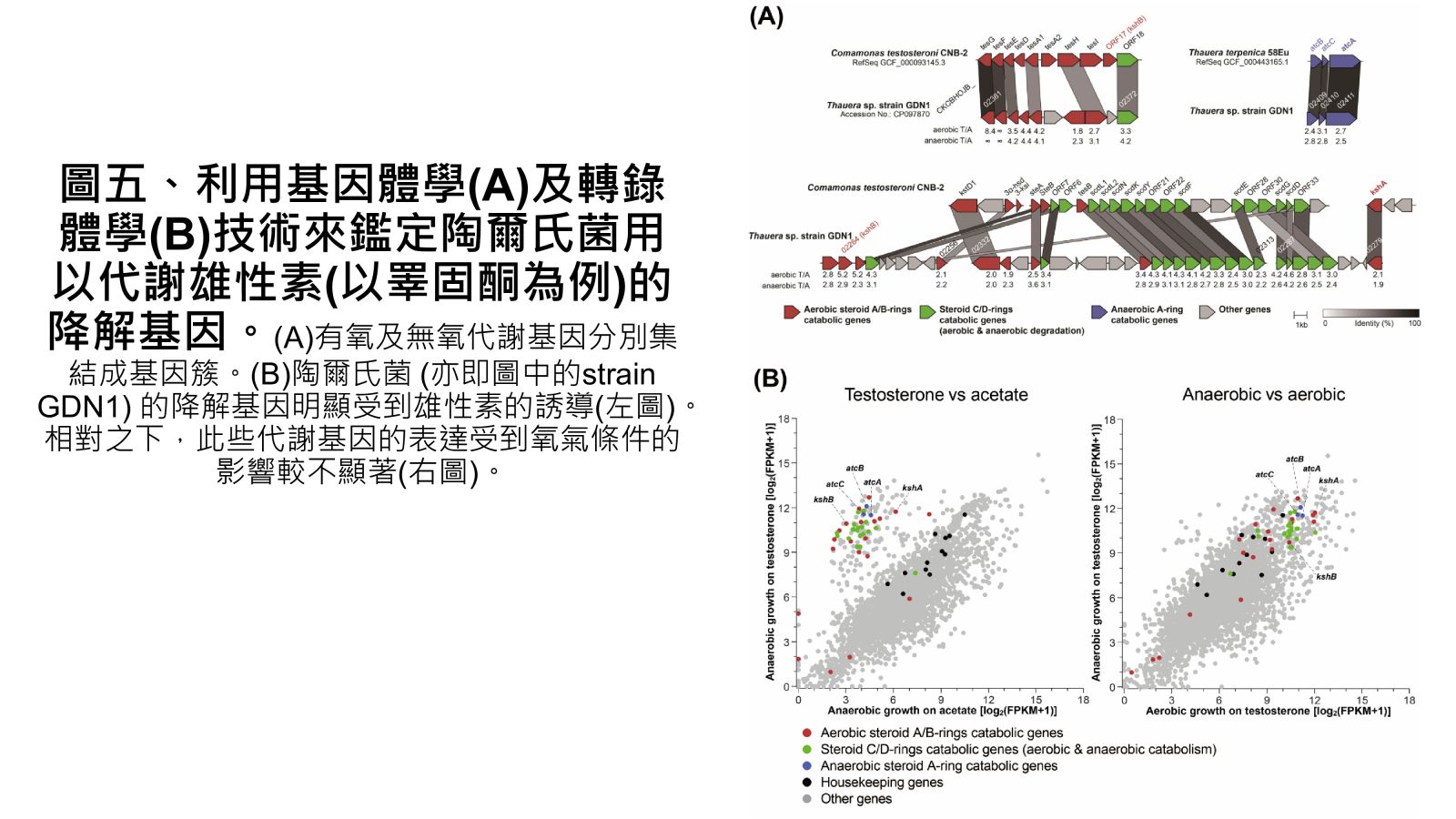
圖五、利用基因體學(A)及轉錄體學(B)技術來鑑定陶爾氏菌用以代謝雄性素(以睪固酮為例)的降解基因。
圖六、餵予陶爾氏菌(亦即圖中的strain GDN1)能影響宿主血清雄性素含量。
圖七、小鼠腸道菌組成之時序性連續分析。
圖八、小鼠腸段定量PCR分析。
圖九、利用超高效液相層析質譜儀來找出並鑑定小鼠腸道中的雄性素代謝物。
表一、自行純化之益生菌菌株之性固醇代謝測試
圖十、益生菌代謝物之雄性素當量活性測試。
圖十一、BRCAS-C3餵予雙氫睪酮小鼠之毛髮恢復情形 。
11用於抗血栓/癌症治療的新型整合蛋白靶向藥物國立成功大學生化所莊偉哲教授發明人:莊偉哲領域:新穎藥物適應症:抗血栓/癌症研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:用於治療抗血栓/癌症的新型整合蛋白靶向藥物
-


抗血小板藥物實驗數據/抗腦瘤藥物實驗數據
12可針對多種癌症遞送抗癌藥物和基因療法之酸鹼應變脫殼和標靶胜肽修飾之奈米技術平台國立陽明交通大學藥理學研究所駱雨利特聘教授發明人:駱雨利領域:劑型開發適應症:癌症研發階段:動物驗證 In vivo validation摘要:目前的癌症治療面臨抗藥性和副作用的挑戰。我們設計新的技術平台,對不同癌症使用適當的miR,以同步調節腫瘤增殖、惡化和抗藥性的多種路徑。我們使用miR和抗腫瘤藥物之奈米劑型,可針對腫瘤部位的酸性pH值產生變化而使得外殼層脫離。接著,這些奈米粒可藉著篩選出的配體與腫瘤上過度表現的受體結合而增加癌標靶辨識性,並利用細胞穿透肽促進腫瘤內吞,而運送標的到細胞核或粒線體,並且可以針對不同癌症使用合適的化療藥物以與miR組合。本研發成果包括三個特色:(1)已優化篩選具有腫瘤表面受體靶向、內體逃離和細胞核/粒線體導向功能的胜肽,與合成pH敏感性高分子,用以製作具有pH應答和多功能胜肽修飾的miR和抗癌藥物奈米粒;(2)可同步抑制腫瘤的致癌、存活、轉移、侵襲或抗藥性等相關機制;(3)以活體荷腫瘤的小鼠,確認奈米劑型所攜帶的抗癌藥物和miR系統,可促進抗腫瘤效應,並且降低血液或器官毒性。總之,開發環境應答型奈米粒,具有可脫離之外殼層和多功能胜肽修飾,並有腫瘤導向和細胞內靶向、同時組合遞送基因和化療藥物到腫瘤部位的特性,故提供了miR和化療藥物合用的新型治療平台,能夠對轉錄後基因調控進行有效的時空控制,並同步抑制腫瘤的多重訊息路徑,所以極具潛力可提高癌症療法的成功率和安全性。
Resistance development and side effects persist as significant challenges in cancer treatment via chemotherapy. Hence, we selected appropriate microRNA (miR) to simultaneously regulate multiple pathways of proliferation, progression, and resistance of tumor cells. The design of miR- and anticancer drug-incorporated nanoparticles can respond to the acidic pH values in tumor sites. The appropriate antineoplastic agents are selected based on characteristics of different cancers. After cleavage of outside polymer shell, these nanoparticles may expose ligands for active targeting to receptors abundant in tumors, and specific cell-penetrating peptides for tumor penetration and internalization, and intracellular localization. These research and development achievements encompass three distinctive features: (1) pH-responsive miR- and anticancer drug-loaded nanoparticles, intricately modified with peptides after optimizing the screening of peptides with functions for tumor targeting, endosomal escape, and nuclear/mitochondrial localization; (2) The concurrent suppression of oncogenesis, survival, metastasis, invasion, and resistance in tumor cells; (3) Enhancement of antitumor efficacy and reduction of organ/blood toxicity using miR- and chemotherapy-loaded pH-sensitive nanoparticles in tumor-bearing mice. Collectively, these nanoparticles boast an exquisite blend of environmental responsiveness, tumor directing, and intracellular targeting, thus delivering gene/chemotherapy combinatorial therapeutics to tumor sites. This exceptional approach enables precise spatiotemporal control of post-transcriptional gene regulation, while effectively inhibiting multiple tumor cell progression pathways, leading to heightened antitumor efficacy and improved safety in cancer therapy.
-
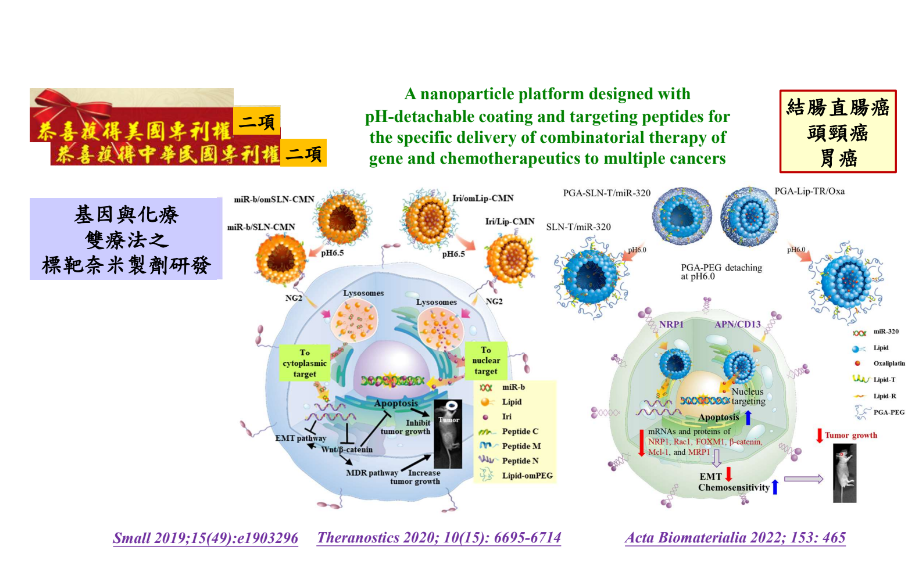
Illustration of the intricate molecular pathways through which nanoparticles, modified with pH-detachable coatings and targeting peptides, enable the precise delivery of combined miRNA and chemotherapy to various cancer types
13Monocyte-Mediated Drug Carriers for Delivering Anti-Cancer Therapeutics國立中興大學程華強助理教授發明人:程華強 李賢明領域:新穎藥物, 劑型開發適應症:cancer, heart failure研發階段:臨床檢體/細胞驗證 Clinical specimen/In vitro validation摘要:(以英文版為主) 最近對過去 10 年報告的各種奈米粒子遞送至不同腫瘤的分析顯示,所施用的奈米粒子中只有 0.7%(中位數)遞送至實體瘤。 因此,許多抗癌奈米顆粒在臨床試驗中失敗也就不足為奇了,因為它們大多有脫靶、治療指數不一致等缺點。 對增強滲透性和保留(EPR)效應的依賴是阻礙許多已開發的奈米顆粒在癌症治療中取得臨床成功的關鍵因素。 然而,很明顯,與重要的正常器官相比,EPR 效應僅使腫瘤組織的遞送增加不到 2 倍。 因此,迫切需要開發能夠以不依賴EPR效應的方式促進藥物標靶治療的抗癌奈米顆粒。 單核細胞募集是癌症發展過程中發生的關鍵先天免疫反應。 進入循環後,循環單核細胞具有最有效到達腫瘤的歸巢能力。 因此,我們決定利用這一病理事件,開發出一種奈米藥物載體,稱為單核細胞介導的藥物載體(MMDC)。 MMDC 靶向並搭便車到被招募到實體瘤的循環單核細胞表面,利用單核細胞作為“超級司機”,將奈米級藥物載體直接運送到實體瘤,而不依賴 EPR 效應。 單核細胞分化為巨噬細胞後,錨定在細胞表面的MMDC立即被細胞吞噬,以達到藥物釋放。 此外,MMDC 對人類單核細胞顯示出強烈的結合親和力,但對人類內皮細胞則不然,這表明藥物載體不會引起不良的凝血效應。 延時影像顯示,我們的 MMDC 可以成功搭上循環單核細胞的便車並滲透到腫瘤微環境中。 更重要的是,標靶是腫瘤特異性的。
A recent analysis of the delivery of a wide range of nanoparticles to different tumors reported over the past ten years revealed that only 0.7% (median) of the nanoparticles administered were delivered to solid tumors. Therefore, it is not unsurprising that many anti-cancer nanoparticles fail clinical trials since most of them suffer from drawbacks such as off-targeting and inconsistency in the therapeutic index. The reliance on the enhanced permeability and retention (EPR) effect is a crucial factor that has hindered the clinical success of many developed nanoparticles in cancer therapy. However, it has become clear that the EPR effects only provide less than 2-fold increases in delivery to tumor tissue compared with critical normal organs. Therefore, there is an urgent need to develop anti-cancer nanoparticles that can facilitate drug-targeted therapy in an EPR-effect-independent manner. Monocyte recruitment is a critical innate immune response that happens during cancer development. Upon entering circulations, the circulating monocytes have the homing capability to reach tumors most efficiently. Therefore, we decided to harness this pathological event and have developed a nano-sized drug carrier called the monocyte-mediated drug carrier (MMDC). The MMDCs target and hitchhike onto the surfaces of circulating monocytes that are being recruited to solid tumors, harnessing the monocytes as “Uber drivers” to carry the nano-sized drug carriers directly to solid tumors without relying on the EPR effect. Upon the differentiation of monocytes into macrophages, the MMDCs anchored on the cell surfaces are immediately phagocytized by the cells, thus achieving drug release. Additionally, the MMDCs show a strong binding affinity for human monocytes but not for human endothelial cells, suggesting the drug carriers would not induce an undesirable clotting effect. Time-lapse images reveal that our MMDCs could successfully hitchhike onto circulating monocytes and infiltrate the tumor microenvironment. More, the targeting was tumor-specific.
-
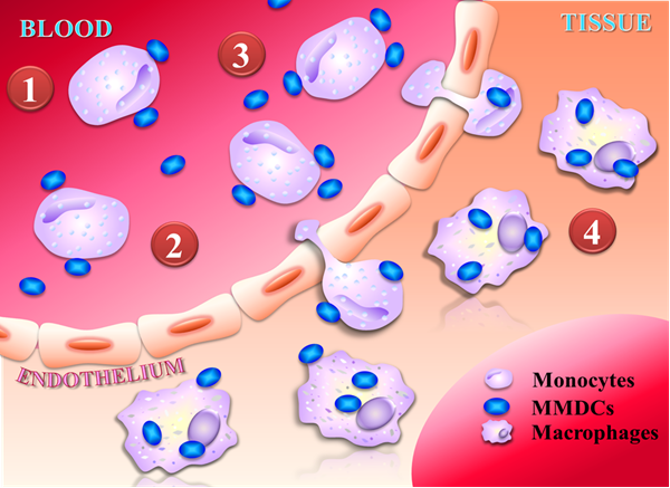
(1) During the progression of chronic diseases such as cancer, heart failure, and renal diseases, monocytes appeared in circulations and displayed strong homing for the targeted sites without relying on the enhanced permeability and retention (EPR) effect.
(2) Our invention is the monocyte-mediated drug carriers (MMDCs), which are designed to hitchhike and anchor on the surfaces of circulating monocytes through monocyte-specific receptors. The MMDCs would remain anchored on the surfaces of monocytes while in circulation since internalization of MMDCs by circulating monocytes could lead to premature drug release.
(3) The monocytes with the MMDCs anchored on their surfaces can still undergo extravasation, carrying the drug carriers near tumors.
(4) After the monocytes have undergone extravasation, the cells differentiate into macrophages, the anchored MMDCs are internalized, and encapsulated drugs are released.14針對漸凍人及相關神經退化疾病之新穎性治療抗體中央研究院基因體研究中心陳韻如研究員發明人:陳韻如領域:新穎藥物適應症:肌萎縮性脊髓側索硬化症(ALS)研發階段:先導藥物最佳化 Lead drug optimization摘要:TDP-43蛋白質沉積出現在數種神經退化性疾病中,包括額顳葉失智症(FTD)、肌萎縮性脊隨側索硬化症(ALS)、和阿茲海默症(AD)。ALS 是一種運動神經元疾病,病人多於發病後 3 至 5 年內離世,目前尚無有效治療方法。ALS治療市場平均每年複合年均增長率估計為12.54%,至2026年全球市場規模至約6億2千萬美元。2014年本團隊於ALS病患腦部檢體中辨識出有毒的TDP-43寡聚體,並生產了具結構專一性的單株小鼠抗體TDP-O,可以辨別錯誤折疊的TDP-43而非正常生理的 TDP-43。本團隊在ALS小鼠模型中證明了它的治療功效。這些抗體已獲美國專利。此專利範圍涵蓋抗體互補決定區(CDR)的必要胺基酸位置,目前已進行人源化,此抗體在 ALS 和其他的TDP-43 蛋白沉積症中具有極大潛力。
TDP-43 proteinopathies were found in several neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Alzheimer’s disease (AD). ALS is a motor neuron disease that leads to death in 3 to 5 years after disease onset. Currently, there is no effective treatment. The global market for ALS is ~$627 million and the compound annual growth rate (CAGR) is 12.54%. Previously, we discovered toxic TDP-43 oligomers and generated conformational-specific monoclonal antibodies, TDP-Os, specifically targeting misfolded TDP-43 but not native TDP-43, and proven the in vivo efficacy of TDP-O9 antibody in an ALS mouse model. The antibodies obtained USA patent covering the conserved residues in the complementarity-determining regions (CDR) regions, which serves as a generic, upper-level patent for future specific patents. The humanized TDP-O antibody targeting misfolded TDP-43 has great therapeutic potential in ALS and other TDP-43 proteinopathies.
-
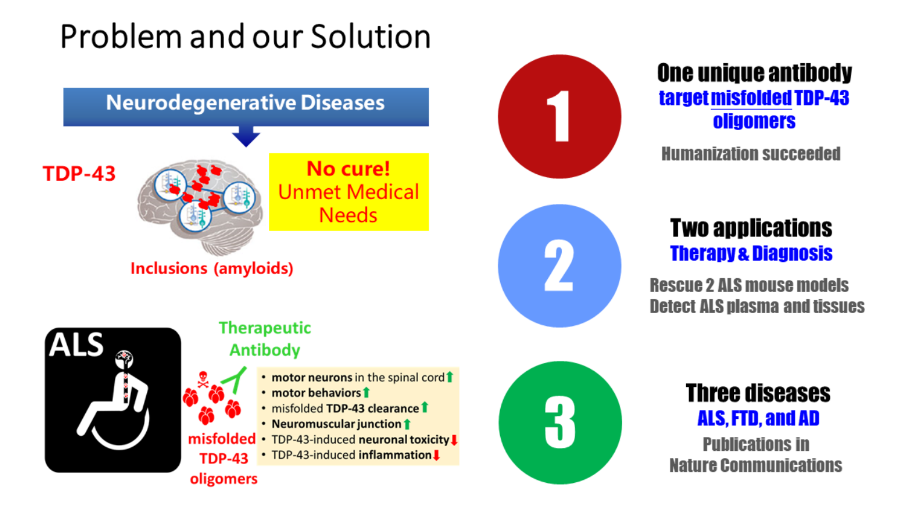
TDP-O抗體在ALS治療以及未來的發展。面對ALS目前缺乏有效藥物的挑戰,我們將專注於ALS指標蛋白TDP-43專一性抗體的研發,從治療和診斷兩個面向著手,在未來力求於ALS,FTD,以及AD三個疾病都能運用。
15開發以ENO1為標靶之抗體新藥中央研究院生醫轉譯研究中心吳漢忠特聘研究員發明人:吳漢忠、李欣蓉、柯鋒翌、斯宜婷、呂瑞旻領域:新穎藥物適應症:肺癌研發階段:動物驗證 In vivo validation摘要:α-enolase (ENO1)是細胞內之醣解酵素,其表現與許多癌症的存活率降低和不良預後顯著相關,然而,ENO1在腫瘤生物學的分子機制尚未完全清楚,目前亦沒有核准上市的抗ENO1抗體藥物。我們近期研究發現,ENO1可經由HGFR和 Wnt訊息傳遞機制增加癌細胞的增殖和侵襲能力。同時,我們運用單株抗體與工程技術製備anti-ENO1人鼠嵌合抗體chENO1-22,證明該抗體能抑制癌細胞生長及侵襲,顯著延長小鼠存活時間,因此,我們推測ENO1是一個具有發展潛力之癌症治療標的。我們成功製作ENO1-22人類化抗體huENO1-22-1,並通過噬菌體人類抗體庫篩選出對抗ENO1人類抗體株hENO1-14,小鼠實驗證明這些抗體具有顯著抑制肺癌細胞轉移與延長存活時間之效果。本計畫將規劃增進hENO1-14之親和力,分析療效與探討其抑癌機制。接續選擇生物活性及療效最好的抗體,完成藥理與毒理試驗,以強化臨床使用之潛力,期望最終開發出以ENO1為標靶之抗體藥物。
-
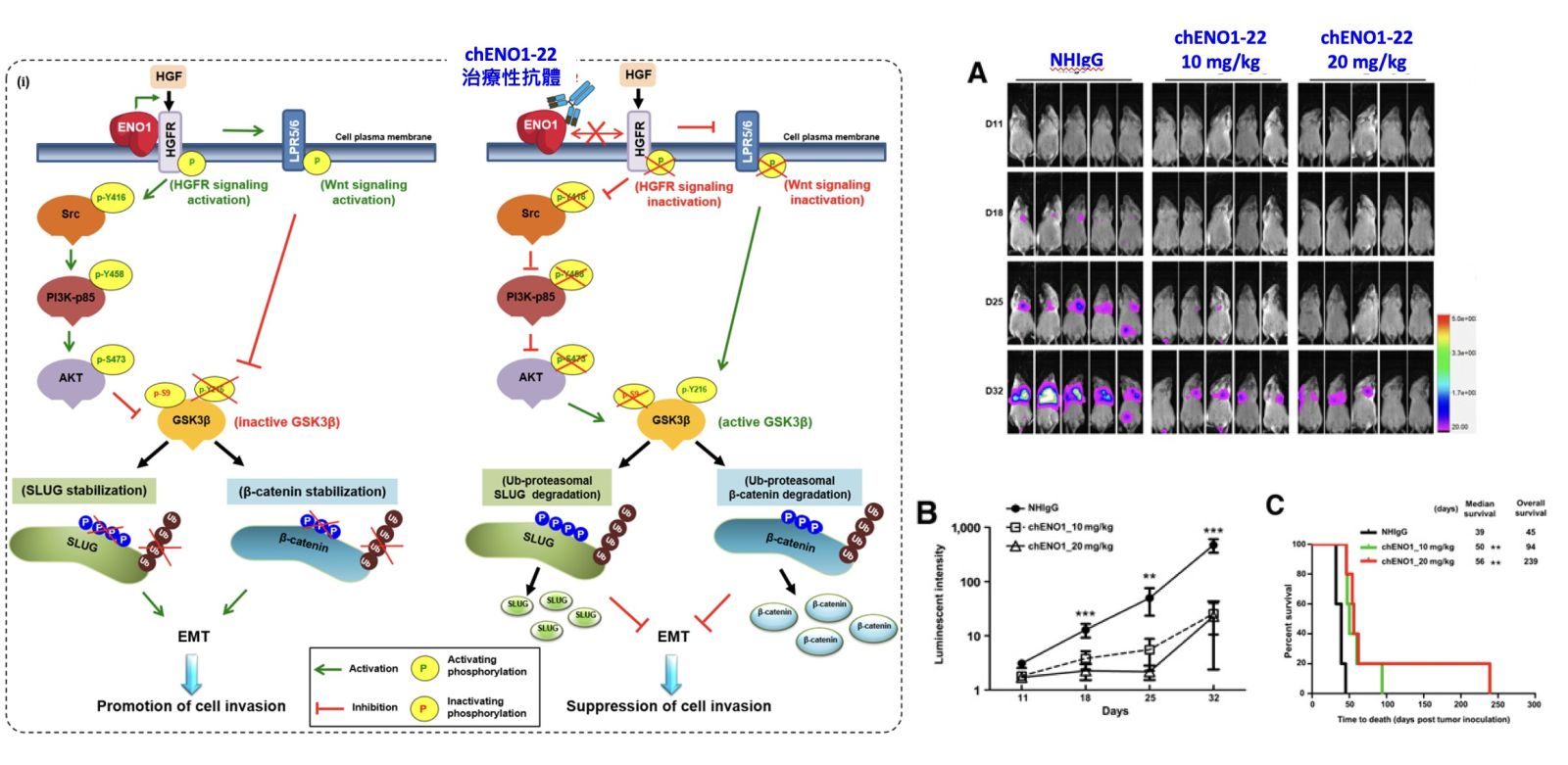
如檔案內容所述
16Establishment of an EpCAM neutralising antibody-mediated combinatorial therapeutics using colorectal cancer patient derived organoids中央研究院生醫轉譯研究中心/細胞與個體生物學研究所吳漢忠特聘研究員發明人:Han-Chung Wu, Sushree Shankar Panda領域:Novel therapeutic strategy適應症:Colorectal cancer研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:Colorectal cancer (CRC) is a devastating disorder with poor prognosis. Despite of strenuous efforts to discover efficacious therapeutics against CRC, the treatment options are limited. Such limitations are due to the presence of mutations such as KRAS and BRAF that mostly exhibit non-responsiveness to the conventional therapeutics. Herein we exploit patient derived organoids (PDOs) as a model system to describe a possible combination therapeutics that may target CRC stem cells including KRAS and BRAF-mutants. Since CRC as well as PDOs contain cancer stem cells (CSCs) in higher numbers that have been implicated in cancer progression, our therapeutic potentially targets CSCs to impede their propagation. In this regard, epithelial cell adhesion molecule (EpCAM) is known to be robustly expressed in CSCs in which case our EpCAM-neutralising antibody EpAb2-6 targets canonical Wnt pathway to supress formation of organoids as well as CSC-derived xenografts. Such effects were pronounced when EpAb2-6 was applied in combination with a porcupine inhibitor. Mechanistically, we show that the extracellular domain of EpCAM (EpEX) functions as an extrinsic cue in the tumor microenvironment to sustain Wnt activity that the CSCs mostly rely on. In this case, EpEX mimics the natural Wnt ligands directly interacting with the Wnt receptors to instigate the signaling. We further identify that activation of Wnt signaling induces the activity of a metalloprotease TACE/ADAM17 that augments cleavage of EpEX enhancing its enrichment in the tumor microenvironment. Therefore, the combinatorial therapeutics depletes such enrichment as well as activation of Wnt ligands to target CSCs. When tested in a patient derive xenograft (PDX) model and various metastatic and orthotopic models, the combinatorial therapeutics exhibited uttermost efficacy prolonging animal survival. Therefore, we establish an EpAb2-6 based combination therapy for the treatment of CRC including KRAS and BRAF-mutants.
-
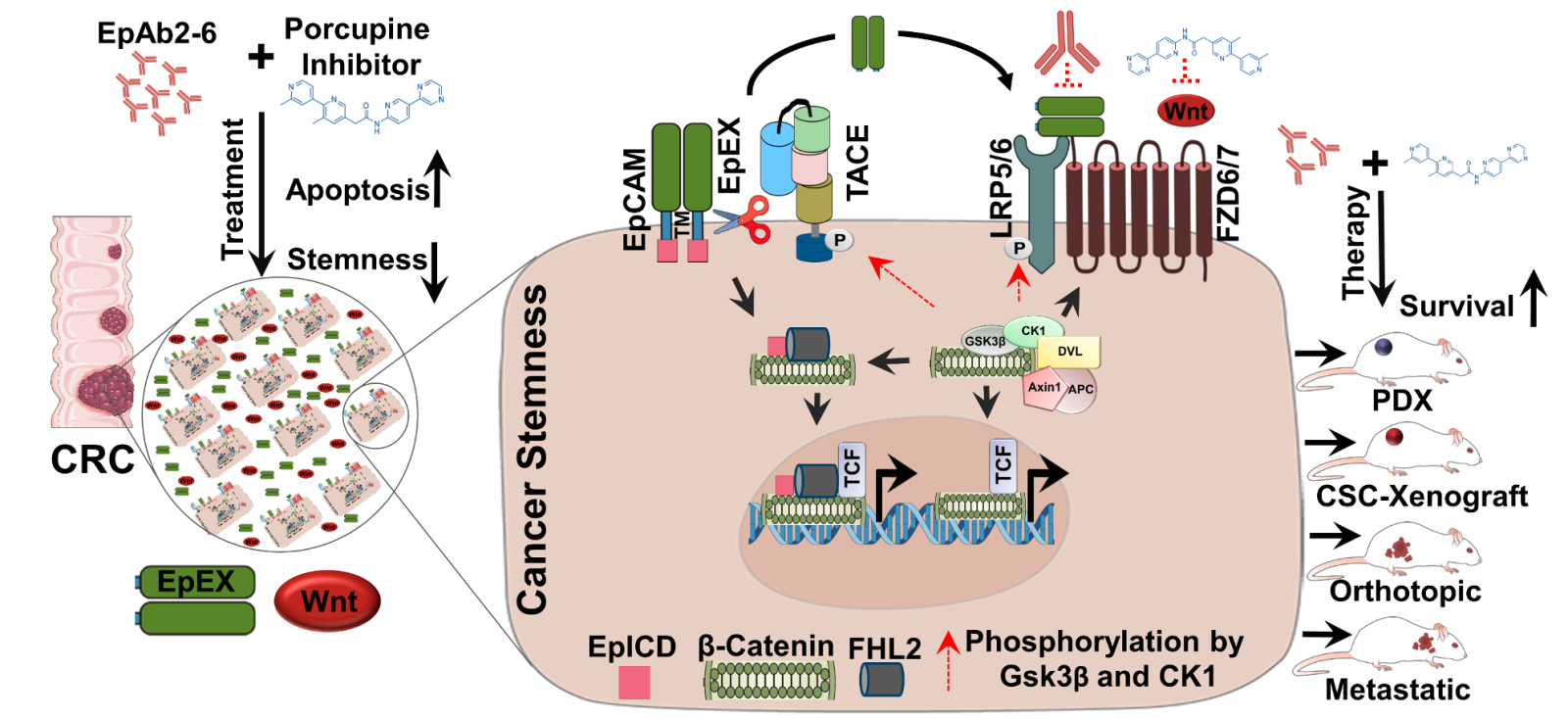
A summary of our discovery how EpAb2-6 and a porcupine inhibitor synergistically inhibit cancer stem cell signaling to exhibit therapeutic efficacy
17研發對抗新型冠狀病毒 Alpha 至 Omicron 之廣效治療性抗體中央研究院生醫轉譯研究中心/細胞與個體生物學研究所吳漢忠特聘研究員發明人:吳漢忠、呂瑞旻、江曉玲、梁剛豪、柯釋涵、陳宛余、林秀亭領域:新穎藥物適應症:COVID-19 infection研發階段:動物驗證 In vivo validation摘要:新型冠狀病毒 (SARS-CoV-2) 自 2019 年底於世界各地爆發疫情,已出現多種高度傳播性且降低疫苗與治療性中和抗體作用之突變株,造成突破性感染。2022 年第四季,Omicron BQ.1.1 與 XBB.1 變異株造成 Eli-Lilly 藥廠研發之中和性抗體 Bebtelovimab 失去效力。目前臨床上無任何中和性抗體能有效抵禦新型變異株的感染,對無法施打疫苗或免疫低下族群造成隱憂。
我們利用人類單一 B 細胞分離技術,挑選 Omicron BA.5 感染痊癒者其對抗棘蛋白 (spike protein) 接受器接合區 (RBD) 之記憶型 B 細胞並進行分離。藉由不同變異株之偽病毒進行中和試驗後,於 hACE2 轉殖鼠中進行確校分析。我們期望此研發之新一代的全人抗體在後疫情時代,提供完整的臨床治療後盾。
-
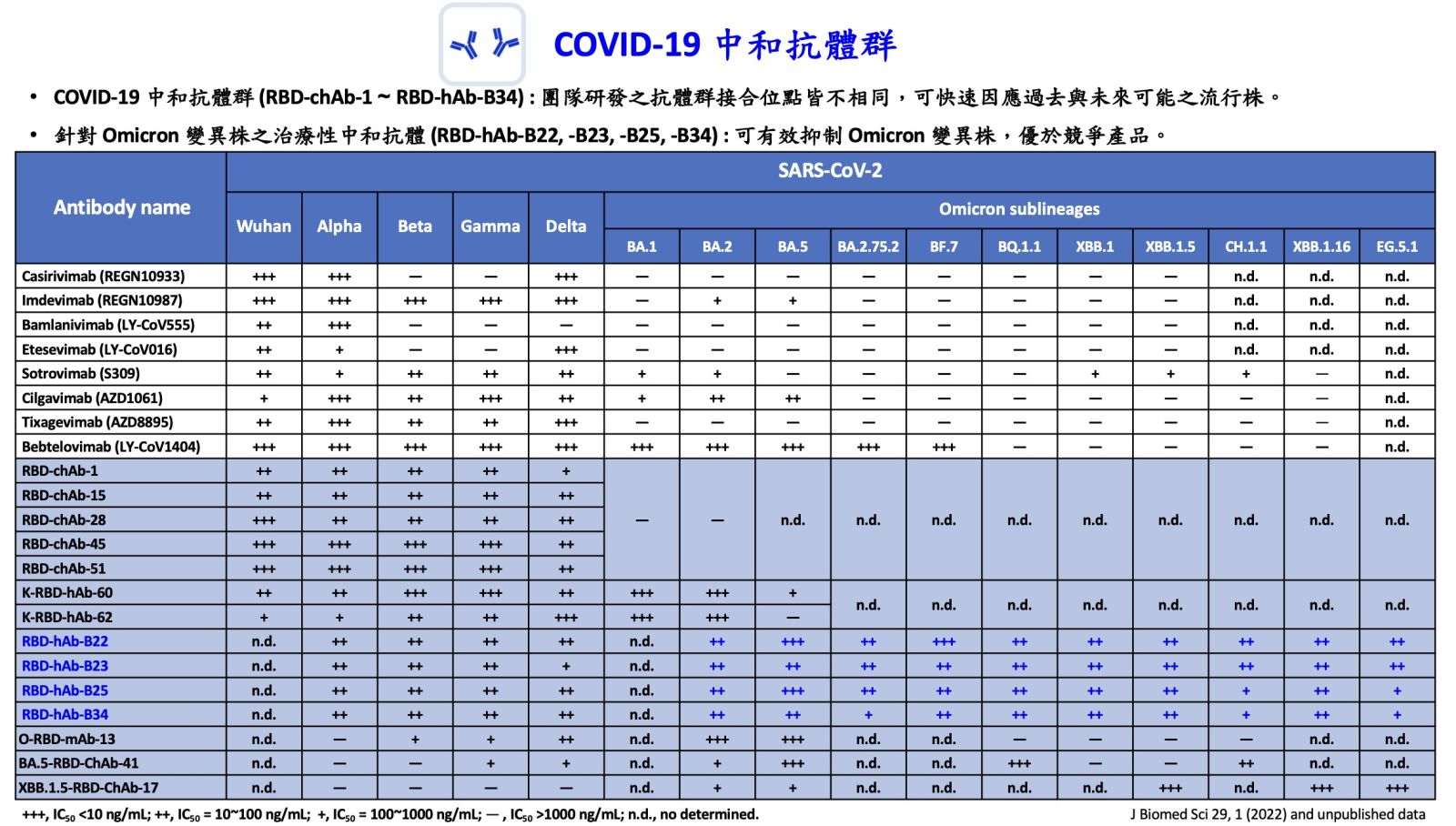
研發對抗新型冠狀病毒 Alpha 至 Omicron 之廣效治療性抗體
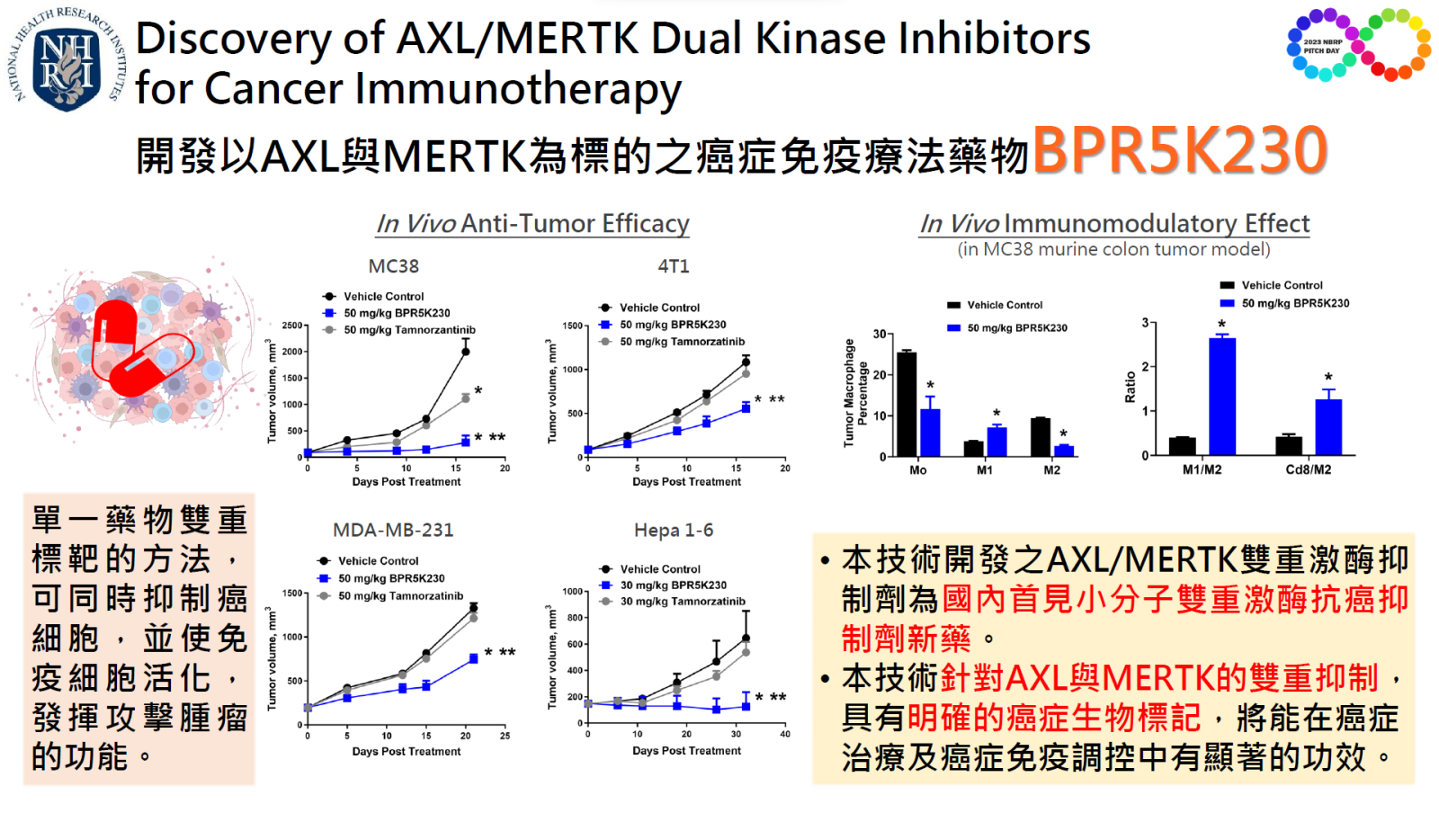
18Discovery of AXL MERTK Dual Kinase Inhibitors for Cancer Immunotherapy國家衛生研究院 生技與藥物研究所謝興邦特聘研究員發明人:謝興邦、顏婉菁、伍素瑩、李慕珺、林文星、柯屹又領域:新穎藥物適應症:三陰性乳癌、肝癌、非小細胞肺癌研發階段:候選藥物/醫材雛型試製造 Pilot production of candidate drug/prototype摘要:AXL 和 MERTK 在腫瘤進展、轉移、耐藥性和免疫逃避中都發揮著重要作用,因此在腫瘤及腫瘤免疫微環境中雙重抑制 AXL 與 MERTK 將增強抗腫瘤功效及抗腫瘤免疫反應。本技術為開發具有高親和力、高度選擇性、低副作用的 AXL 與 MERTK 新穎雙重激酶抗癌小分子抑制劑。我們運用國衛院生藥所專利保護之特有激酶特異性小分子化合物資料庫鑑定出 BPR5K230,其具有 AXL 與 MERTK 激酶雙重抑制活性及對 TYRO3 的選擇性,且優於競爭藥物 tamnorzatinib。作為一免疫治療劑,BPR5K230 可減少 4 倍腫瘤內的 M2 腫瘤相關巨噬細胞 (TAM),並在脾臟中增加 3 倍的效應 T 細胞。在動物試驗中,BPR5K230 單獨或與免疫檢查點抑制劑合併使用可產生體內抗腫瘤功效。其抗癌活性優於競爭藥物 tamnorzatinib。作為一標靶藥物 BPR5K230 對 AXL 與 MERTK 的雙重抑制抗腫瘤功效優於 AXL 或 MERTK 單一標靶藥物抑制療法,驗證了 BPR5K230 對 AXL 和 MERTK 的雙重抑制。
AXL and MERTK are members of TAM (TYRO3, AXL and MERTK) receptor tyrosine kinases. Both AXL and MERTK play important roles in tumor progression, metastasis, drug resistance and immune evasion. Thus, dual AXL and MERTK inhibition in the tumor and tumor immune microenvironment would enhance anti-tumor efficacy and boost anti-tumor immune responses. Utilizing our proprietary small molecule tyrosine kinase inhibitors compound library, we identified BPR5K230 with potent AXL and MERTK kinase inhibitory activities and selectivity over TYRO3. BPR5K230 produced in vivo anti-tumor efficacy, alone or in combination with immune checkpoint inhibitors. In multiple preclinical models evaluated, the anti-tumor effect of BPR5K230 was more efficacious than the clinical stage agent tamnorzatinib. As an immunotherapeutic agent, BPR5K230 decreased 4-fold M2 tumor-associated macrophages (TAM) in the tumor and increased 3-fold effector T cells in the spleen. As a molecular-targeted agent, BPR5K230 produced greater anti-tumor efficacy than either AXL or MERTK mono-targeted agent used alone, verifying BPR5K230’s dual AXL and MERTK inhibitory activities. Provisional patent has been filed.
 31治療失明之新型細胞治療策略中央研究院基因體研究中心呂仁副研究員發明人:呂仁、蔡榮坤、李岳章、賴培倫、賴倩瑩領域:新穎藥物適應症:黃斑部病變、夜盲症研發階段:申請試驗用藥物/新醫材(試劑) Investigational New Drug (IND)/ Investigational Device Exemption (IDE) application摘要:
31治療失明之新型細胞治療策略中央研究院基因體研究中心呂仁副研究員發明人:呂仁、蔡榮坤、李岳章、賴培倫、賴倩瑩領域:新穎藥物適應症:黃斑部病變、夜盲症研發階段:申請試驗用藥物/新醫材(試劑) Investigational New Drug (IND)/ Investigational Device Exemption (IDE) application摘要:流行病學調查顯示,失明是一種令人恐懼的疾病,其對人類的影響超越了阿茲海默症和癌症。根據世界衛生組織的數據,全球約有10%的失明病例是由感光細胞退化引起的。我們的研究專注於解決感光細胞退化疾病所帶來的挑戰,市場價值預估將在2030年達到200億美元。本技術利用小分子藥物將人類纖維母細胞誘導為視網膜前驅細胞,轉化效率高達42.8%,僅需5天的時間。這項技術在臨床應用中具有便利性和低成本的優勢。該細胞轉化過程無需基因改造或病毒介入,在動物實驗中已驗證了其治療效果,並且沒有生成腫瘤的風險。與我們的主要競爭對手相比,他們只能拯救黑白視覺,而我們的產品可能能夠恢復黑白和彩色視覺,為患者提供更全面的視覺體驗。我們的技術在醫療市場上具有競爭優勢和發展潛力,特別是在黃斑部病變、糖尿病視網膜病變、和視網膜色素變性(罕見疾病)。我們將與醫療機構和製藥公司建立合作夥伴關係,實現視網膜前驅細胞在細胞治療中的應用,並為感光細胞退化患者提供新的治療選擇。
Epidemiological investigations have revealed that blindness is an extremely frightening disease, surpassing Alzheimer's and cancer in its impact on humanity. According to data from the World Health Organization, approximately 10% of global blindness cases are caused by degeneration of photoreceptor cells. Our research addresses the challenges posed by photoreceptor degeneration, with a market value reaching 20 billion dollars in 2030. Our technique involves utilizing small molecules to induce human fibroblast cells into retinal progenitor cells. We achieved a significant conversion efficiency of up to 42.8% in just 5 days. This approach offers the advantages of convenience and cost reduction in clinical applications. The cell conversion process does not require genetic modification or viral intervention, and its therapeutic efficacy has been validated in animal experiments with no risk of tumor formation. In contrast to our major competitor who can only rescue black-and-white vision, our product might be able to restore both black-and-white vision and color vision, will provide patients with a more comprehensive visual experience. Our technology has competitive advantages and great development potential in medical markets such as macular degeneration, diabetic retinopathy, and retinitis pigmentosa (a rare disease). We will establish partnerships with medical institutions and pharmaceutical companies to realize the application of retinal progenitor cells in cell therapy and provide new treatment options for patients with photoreceptor degeneration.
-

Our technology involves using small molecules to directly convert human somatic cells into retinal progenitor cells in as little as 5 days, with the fastest current conversion method taking 10 days. This approach has significantly higher visual resolution, up to 20,000 times that of known competitors. Unlike previous studies that only restored black-and-white vision, our chemically induced retinal progenitor cells (CiRPCs) can differentiate into rods and cones, thereby restoring monochromatic and color vision, while leading competitors can only restore black-and-white vision. This breakthrough provides patients with a significantly improved visual experience. Animal trials have demonstrated its effectiveness in restoring light sensitivity in blind rats.
32ADAM9抑制劑作為新穎抗胰臟癌藥物中國醫藥大學佘玉萍教授發明人:佘玉萍/Yuh-Pyng Sher領域:新穎藥物適應症:胰臟癌/Pancreatic cancer研發階段:候選藥物/醫材雛型臨床前試驗 Preclinical trials摘要:胰臟癌難根治,90% 攜帶 KRAS 突變,其5 年存活率低於 10%。阻斷 KRAS 表達和活性可有效對抗胰腺癌。然而,胰臟癌存在十幾種 KRAS 突變類型,且多突變型共存在於腫瘤內,增加治療困難。我們開發的 ADAM9 抑制劑作為泛 KRAS 抑制劑,可有效降低腫瘤中的高 KRAS 活性,並促進多種 KRAS 突變體和野生型的 KRAS 蛋白降解。胰臟癌動物模型證明了 ADAM9 抑制劑具抑制胰臟癌進展的潛力,與Gemcitabine併用,強化抗癌作用,人類胰腺癌動物模型中消除了 70% 的腫瘤,安全性高。可減少臨床胰臟腫瘤 KRAS 蛋白,顯示其臨床應用潛力。我們已獲得 ADAM9 抑制劑在癌症治療中的核心結構和免疫調節應用專利。因此,此 ADAM9 抑制劑具有作為術後輔助治療,減輕腫瘤復發,可有效控制疾病進展,提高患者的生活品質。
Pancreatic cancer is a devastating disease, with a 5-year survival rate of under 10% for nearly 90% of patients who carry the KRAS mutation. Efforts to combat pancreatic cancer have shown promise in blocking KRAS expression and activity. However, the complexity arises from over a dozen KRAS mutant types in pancreatic cancer patients, and multiple mutants often co-occur within a single tumor. This diversity poses challenges for targeting KRAS, as current drugs can only address specific mutations, failing to tackle the multiple KRAS mutations found in tumors.
Our team has risen to this challenge; our developed ADAM9 inhibitors as pan-KRAS inhibitors effectively reduce high KRAS activity in tumors and promote KRAS protein degradation across multiple KRAS mutant and wild types. Preclinical studies utilizing pancreatic cancer animal models demonstrate the potential of ADAM9 inhibitors to curb pancreatic cancer progression. Their combination with the frontline drug Gemcitabine enhances anticancer effects, eradicating 70% of tumors in human patient-derived pancreatic cancer animal models. Importantly, these treatments exhibit no toxicity in the liver, kidney, or blood in preclinical cancer animal models. Furthermore, their effects of reducing KRAS proteins in clinical pancreatic tumors underscores their clinical application potential.
We have filed core structure and immune modulation application patents for ADAM9 inhibitors in cancer treatment. To safeguard intellectual property rights, we intend to file patents for drug formulation and diagnostic tools to evaluate treatment responses. Consequently, our ADAM9 inhibitors hold therapeutic promise as adjuvant therapy post-surgery to mitigate tumor recurrence. Combined with the clinical drug Gemcitabine, they can effectively manage disease progression, enhancing the quality of life for pancreatic cancer patients. Our ultimate goal is to empower cancer patients by averting tumor recurrence and extending their lives.-
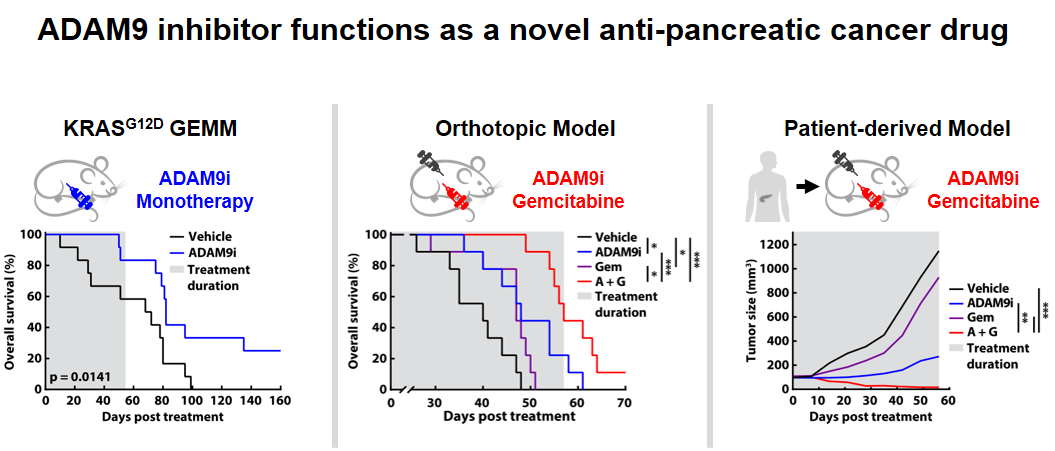
Multiple preclinical animal models fully support that ADAM9 inhibitors reduce pancreatic cancer progression. Combined with Gemcitabine, it can achieve the effect of tumor eradication in pancreatic cancer treatment.
33智慧化創新臨床前新藥研發平台臺北醫學大學 醫學科技學院潘秀玲院長發明人:潘秀玲、許凱程領域:新穎藥物, 人工智慧新藥開發適應症:難治型癌症與神經退化性疾病研發階段:候選藥物/醫材雛型試製造 Pilot production of candidate drug/prototype摘要:智慧化創新臨床前新藥研發平台是擁有三個高準確率計算模型的藥物開發工具。提供設計新穎抑制劑、篩選蛋白標的、預測細胞毒性、預測ADMET及細胞/動物實驗驗證的服務。可縮減臨床前研發時程至2年內,提高14倍的研發成功率。目前本團隊已應用此平台於3年內開發出25個高潛力新穎小分子抑制劑研發專案,且多數在臨床上無競爭產品,並已服務17個國內客戶。
Focusing on oncology and neurodegenerative diseases with unmet needs, our team developed an end-to-end preclinical AI platform depending on our >25 years of drug R&D and chemical synthesis experiences. We utilize our professionals to build this innovative AI platform, which is capable of swiftly generating patentable structures with enhanced activity in just 17 days. Furthermore, it accelerates preclinical development to 2-3 years. Importantly, all compounds generated by our AI platform are highly synthesizable and can be produced in a single step. This results in reduced costs, shorter timelines, and improved success rates, offering an effective solution.
Having successfully developed 25 high-quality preclinical assets (IC50<10nM) through our AI platform and having served 17 customers in Taiwan, including academia and biotech, we are excited to share this journey with you.-
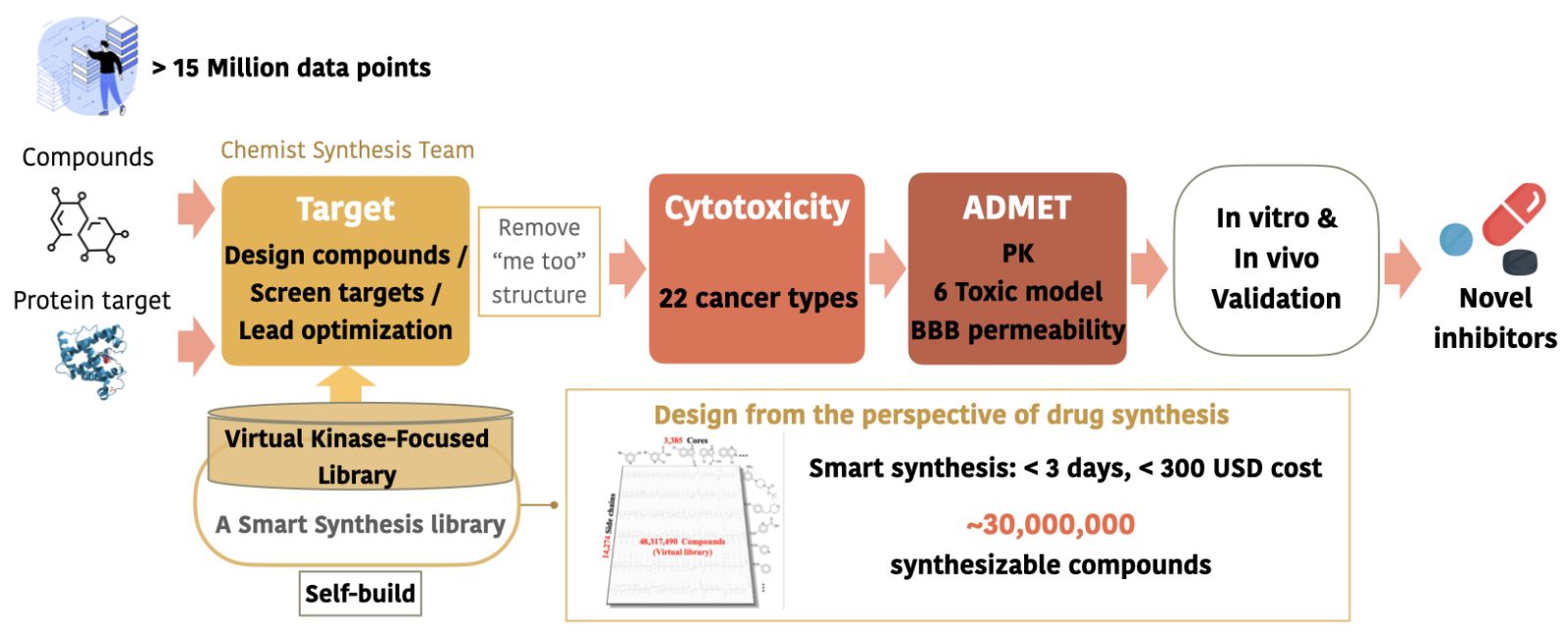
平台擁有超過千萬筆經處理之正確資料,搭配團隊自建的虛擬化合物資料庫,可經由3個主要模型快速研發、預測出高潛力 first-in-class 新藥產品。
34雙價腸病毒疫苗及季節性流感VLP疫苗國家衛生研究院李敏西研究員發明人:李敏西領域:新穎藥物適應症:腸病毒感染症及季節性流感研發階段:動物驗證 In vivo validation摘要:縱觀近代疫苗開發的成功案例,諸多仰賴公衛法規基礎架構及醫學研究,且需搭配委託研究機構及委託開發生產機構等提供周邊服務,通常由中小型疫苗公司進行第 1-2期臨床試驗,然後由疫苗廠接手第 3期臨床試驗及上市行銷。台灣目前已有兩家疫苗廠,最好需有 5-10家中小型疫苗公司專注第一、二期疫苗臨床試驗,但目前小於5家的中小型疫苗公司,需要更多疫苗新創公司來彌補這個缺口。國衛院李敏西研究員擁有國內外 30年以上疫苗研發經驗,加入國衛院疫苗研發團隊後,曾參與數個國家型計劃,建立疫苗團隊開發腸病毒和流感疫苗,為了強化台灣疫苗研發能量,擬成立疫苗新創公司,將雙價腸病毒疫苗及季節性流感類病毒顆粒 (VLP)疫苗推進到臨床試驗。
There are currently two vaccine factories in Taiwan. Therefore, it is best to have 5-10 small and medium-sized vaccine companies focusing on Phase 1 and 2 clinical trials. However, there are currently less than 5 small and medium-sized vaccine companies, and more vaccine start-up companies are needed to make up for this gap. Dr Min-Shi Lee has more than 30 years of experience in vaccine research and development. He has participated in vaccine research and development in a vaccine company in California, USA for 6 years. After joining NHRI in 2005, he has participated on several national projects working on vaccine development against enterovirus and influenza H5N1, H7N9 and seasonal influenza. To strengthen Taiwan’s vaccine research and development capabilities, Dr Min-Shi Lee’s group has established a vaccine start-up company for developing bivalent enterovirus vaccines and seasonal influenza virus-like particle (VLP) vaccines and bringing these vaccine candidates into clinical trials.
-
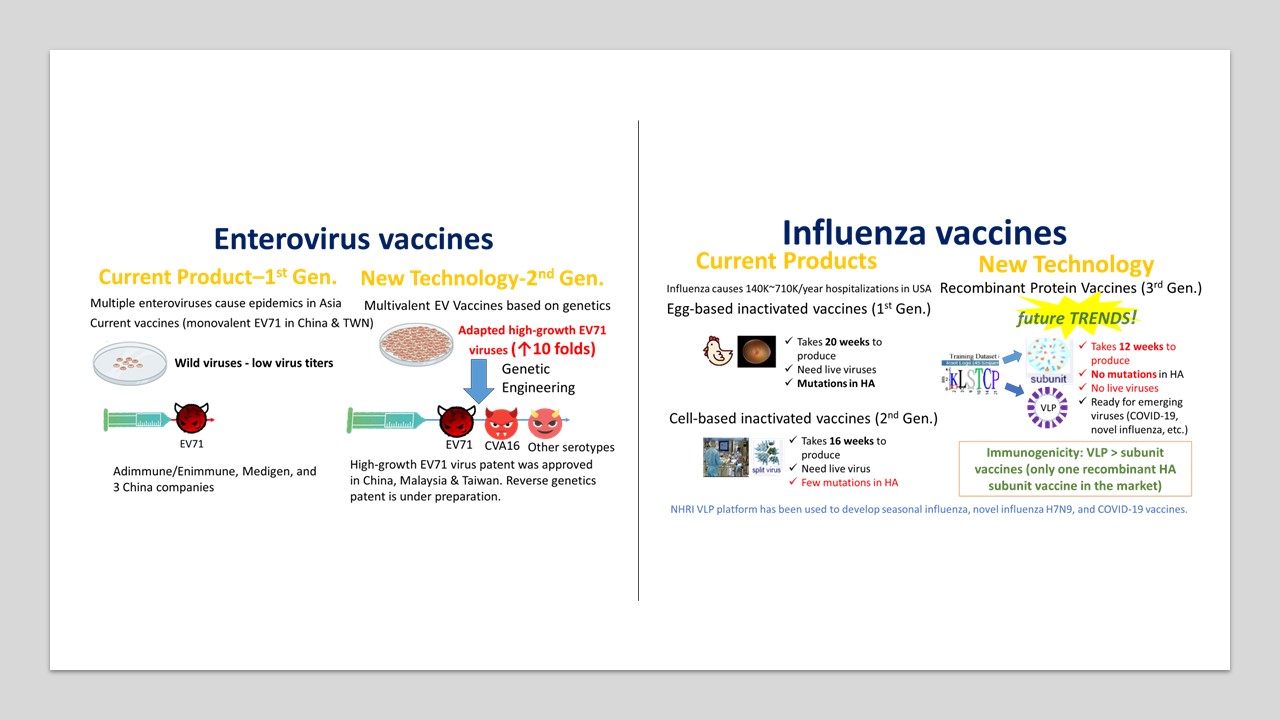
腸病毒疫苗: 現今發展中的多價腸病毒疫苗主要分為兩大類,類病毒顆粒 (VLP) 以及去活化病毒(Inactivated virus),本團隊結合高成長疫苗株及基因工程技術,可大幅降低生產成本,具市場競爭力,並已馴化出高成長病毒株,和野生病毒相比病毒效價提高10倍。
季節性流感疫苗: 現今上市的季節性流感疫苗主要分為三大類:雞胚蛋疫苗(第一代)、狗腎細胞培養疫苗(第二代) 、重組HA蛋白疫苗(第三代)。雞胚蛋疫苗生產缺乏彈性且易產生抗原變異,將逐漸被淘汰;狗腎細胞疫苗需要繁殖病毒難以因應新興流感病毒;重組HA蛋白疫苗致免疫力較差,本團隊開發之流感VLP疫苗只需要第一級生物安全設施操作,生產效率高,且致免疫力高於重組HA蛋白疫苗,除了可生產季節性流感疫苗,也可有效因應新興流感病毒及其他新興傳染病。35具EGFR表現活化相關癌症之治療新靶點及其胜肽藥物國立臺灣大學 生物技術研究中心、臺大植微系沈湯龍主任兼教授發明人:沈湯龍 / Tang-Long Shen , 戴佑玲 / Yu-Ling Tai領域:新穎藥物適應症:EGFR活化參與相關癌症包含乳癌、結腸癌、皮膚癌、肺癌或胃癌 / Related cancers involving EGFR activation, including breast cancer, colon cancer, skin cancer, lung cancer or gastric cancer研發階段:動物驗證 In vivo validation摘要:根據美國食品與藥物管理局定義,胜肽藥物係指胺基酸數目在40個以下的分子,本發明之FAK/11a.a.或FAK/25a.a.胜肽藥物,其治療靶點在FAK-Tyr397自我磷酸化位點前的11個胺基酸區域作為負責β4整聯蛋白結合,本發明為短鏈胜肽藥物,具備生物藥對專一性靶點的強效結合力且毒性更低,亦具有小分子藥物進入細胞的能力。本發明已取得臺灣及美國專利權保護,可應用於EGFR活化參與相關癌症包含乳癌、結腸癌、皮膚癌、肺癌或胃癌,並已完成結腸癌及乳癌相關細胞及動物驗證實驗。本發明為短鏈胜肽藥物可以循新藥申請(New Drug Application, NDA)規定提交,在製程成本上相較蛋白質候選藥物等生物製劑更具優勢,成為「負擔得起的治療」,可預見本發明的First-to-Patent新治療靶點及其First-in-class胜肽藥物極具臨床應用市場潛力。
FDA considers any amino-acid polymer composed of 40 or fewer amino acids to be a peptide rather than a protein. Our invention includes a peptide drug with FAK/11a.a. or FAK/25a.a. for the novel therapeutic target, which we found that an 11-amino-acid region ahead of the FAK-Tyr397 autophosphorylation site is responsible for β4 integrin in interaction with FAK in regulating various cancer progression including metastasis. Our short-chain peptide drug is similar to other biopharmaceuticals conferred with a strong binding to the corresponding specific target but with lower toxicity, as well as having a higher ability into cells like small molecule drugs. Our peptide drug sequence and its therapeutic target have been successfully granted the patent rights in Taiwan and the United States, and can be applied to related cancers involving EGFR activation, including breast cancer, colon cancer, skin cancer, lung cancer or gastric cancer, and we have completed relevant cell and animal verification experiments on colon cancer and breast cancer. Our short-chain peptide drug has the advantage of low manufacturing cost and can be submitted in accordance with New Drug Application (NDA) regulations. Compared with biologics such as protein drugs in terms of manufacturing costs, it has become an "affordable treatment". Accordingly, our First-to-Patent therapeutic target for EGFR-relevant cancer and the First-in-class peptide drug therefore will have more potential applications on the clinical market.
-
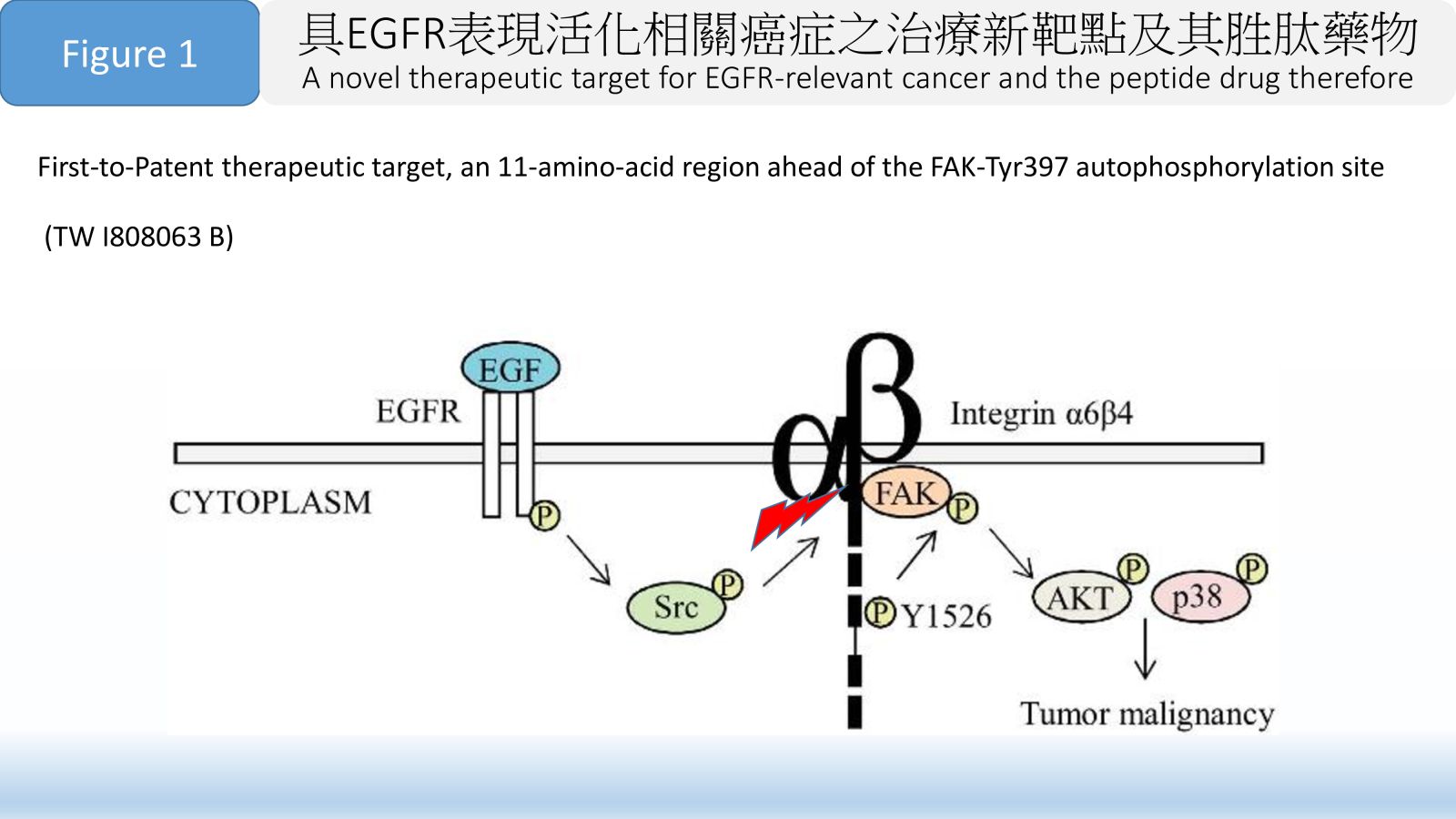
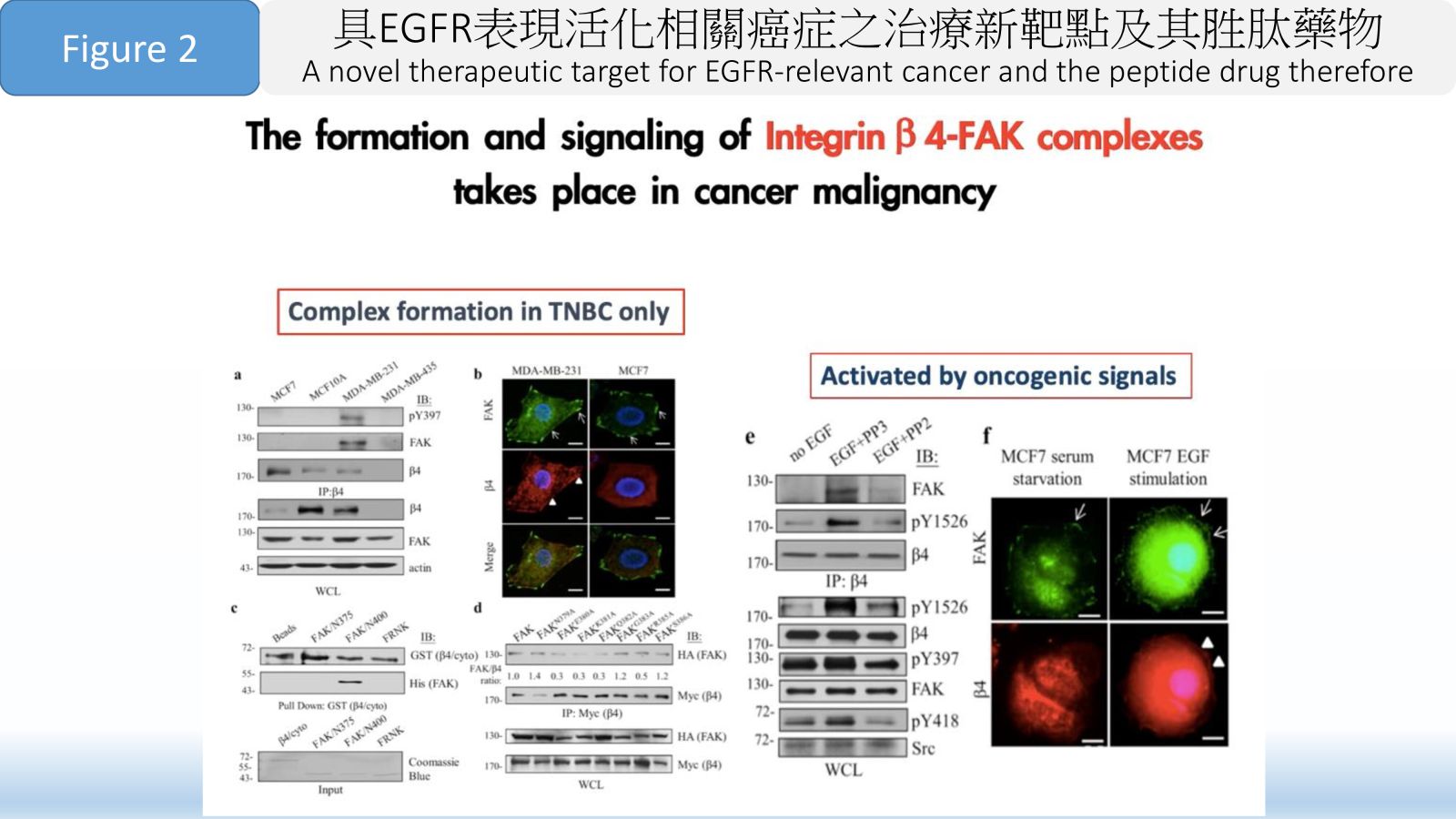
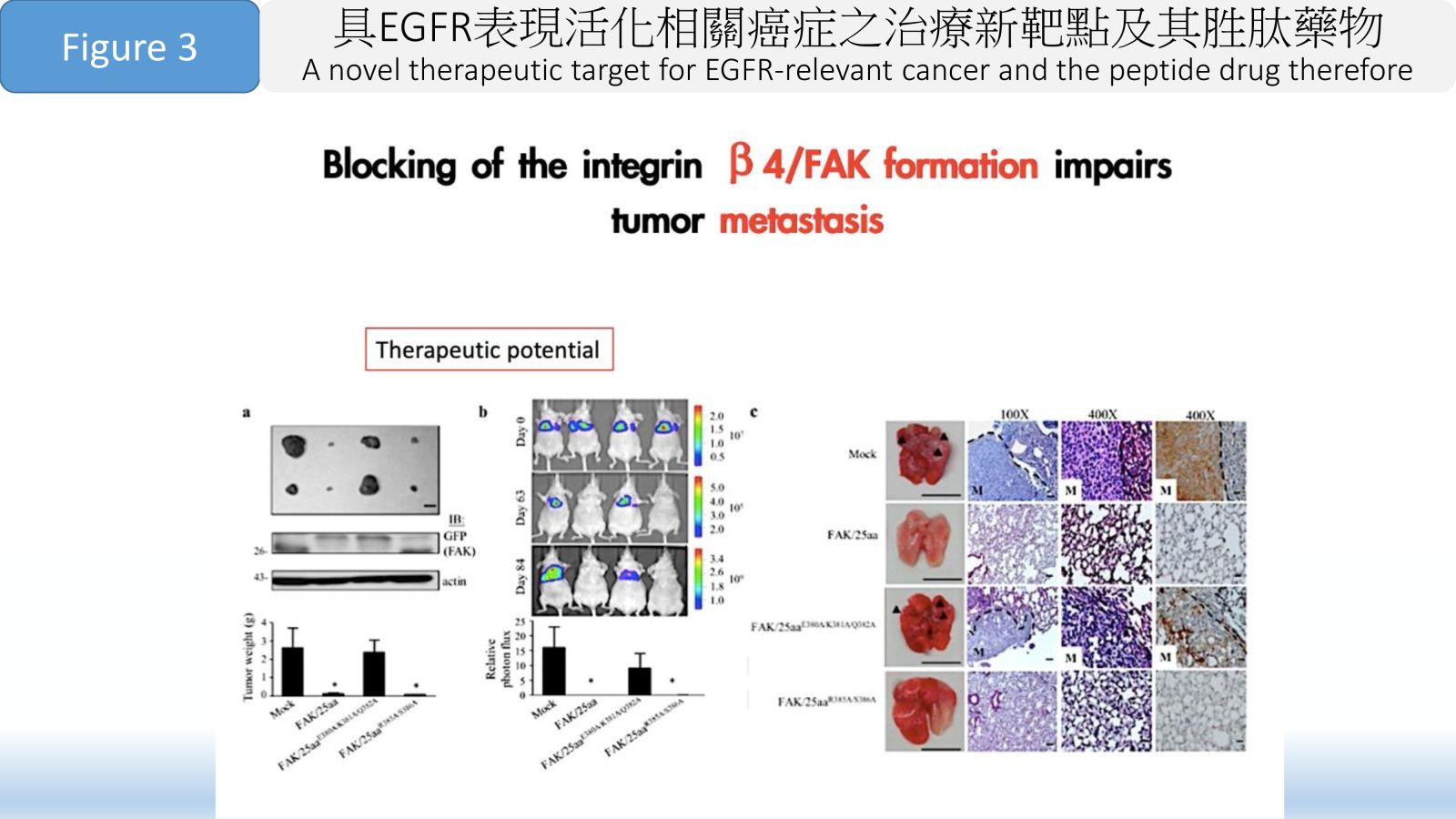
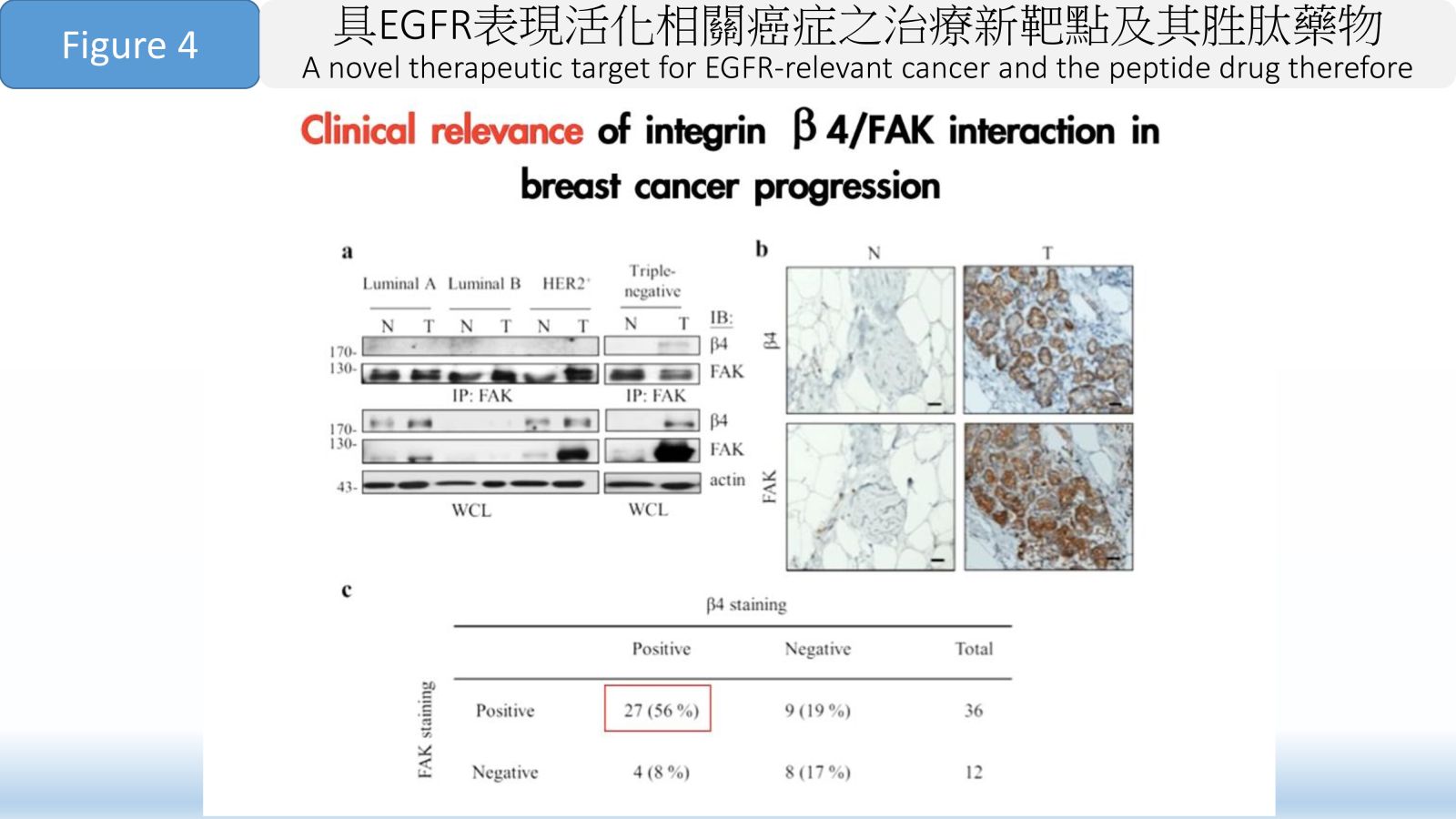
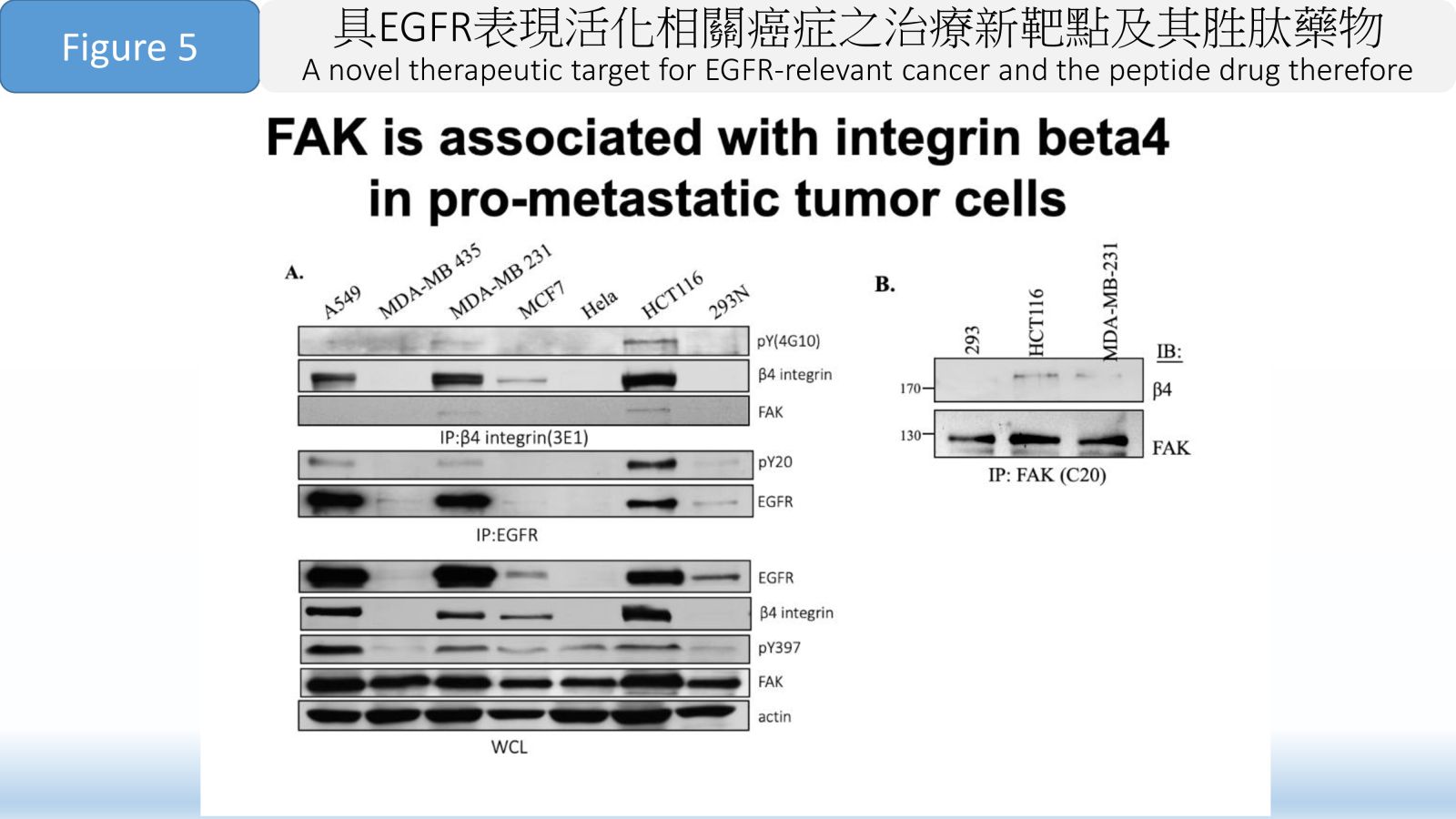
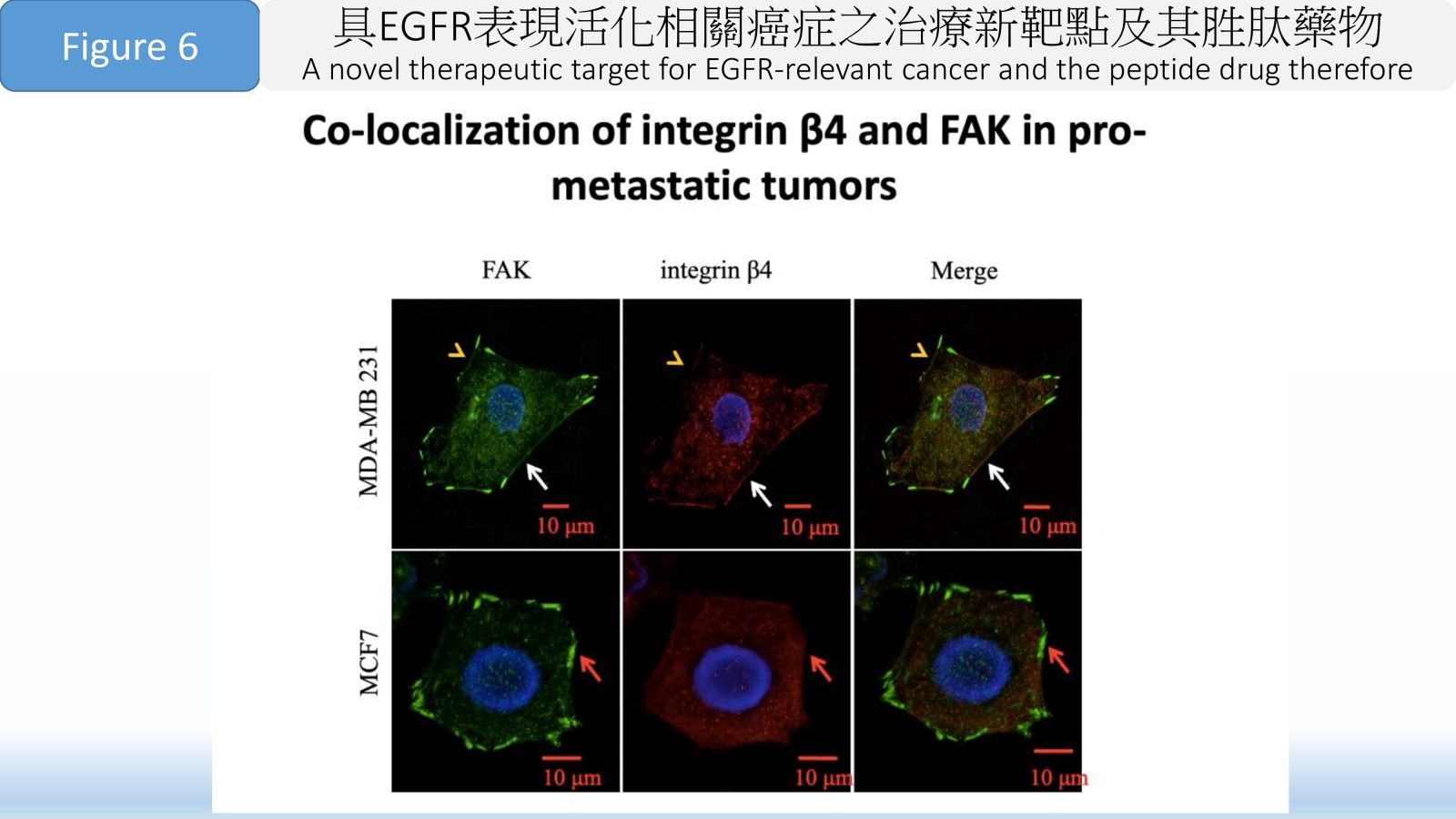
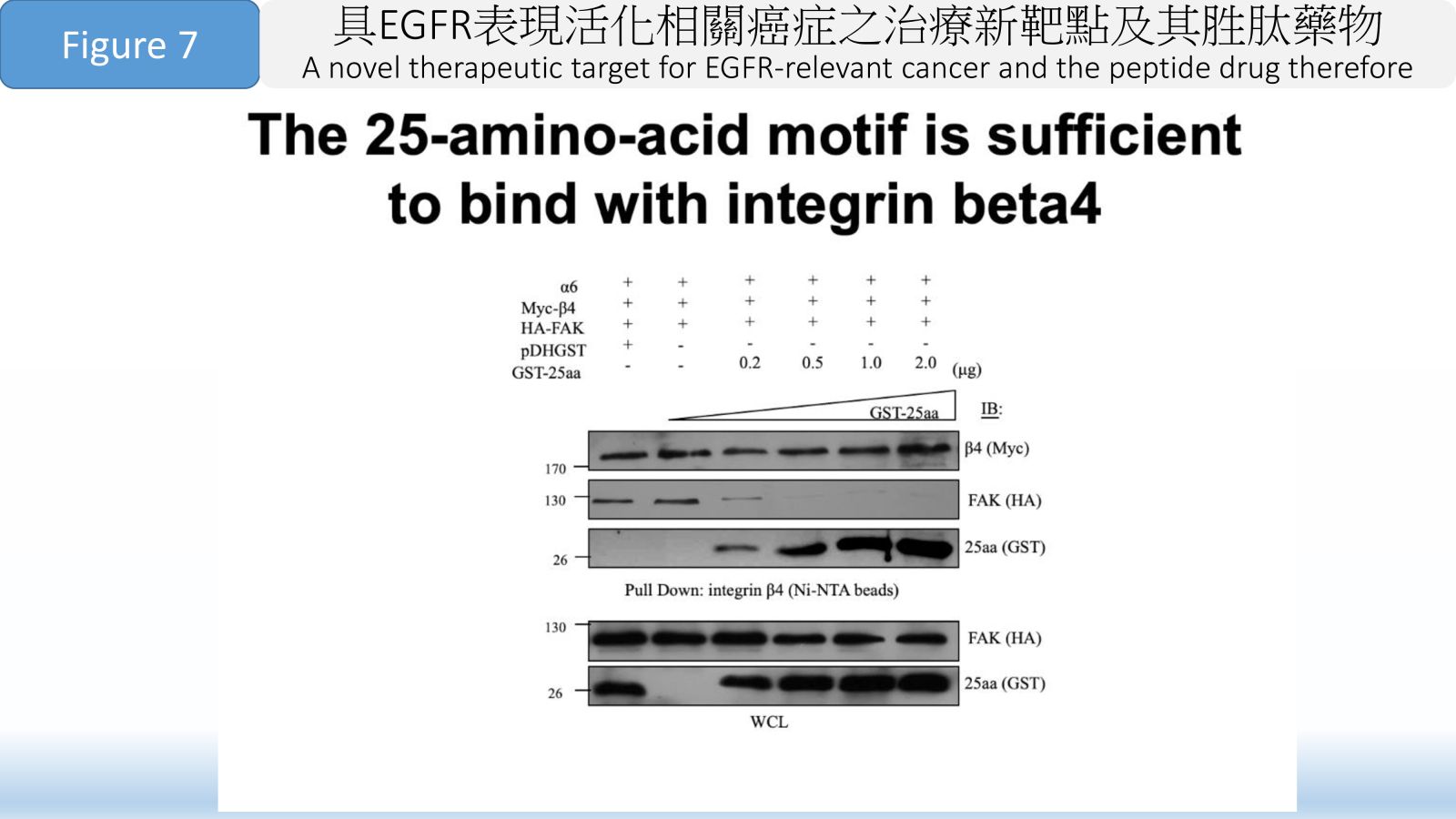
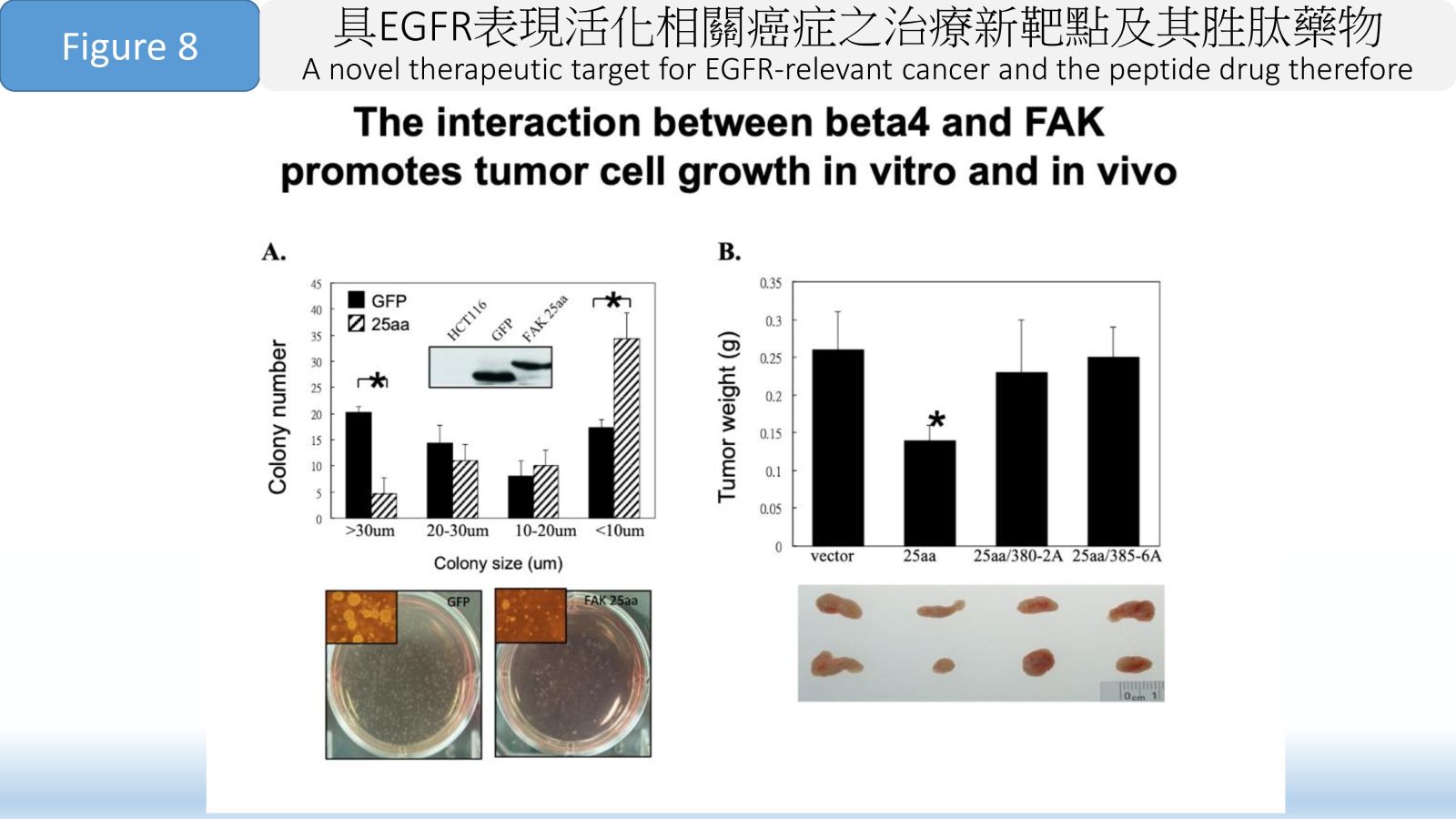
Figure 1 First-to-Patent therapeutic target, an 11-amino-acid region ahead of the FAK-Tyr397 autophosphorylation site. (TW I808063 B)
Figure 2 The formation and signaling of Integrin β 4-FAK complexes takes place in cancer malignancy.
Figure 3 Blocking of the integrin β 4/FAK formation impairs tumor metastasis.
Figure 4 Clinical relevance of integrin β 4/FAK interaction in breast cancer progression.
Figure 5 FAK is associated with integrin beta4 in pro-metastatic tumor cells.
Figure 6 Co-localization of integrin β4 and FAK in pro-metastatic tumors.
Figure 7 The 25-amino-acid motif is sufficient to bind with integrin beta4
Figure 8 The interaction between beta4 and FAK promotes tumor cell growth in vitro and in vivo.36廣效奈米藥物-醣苷轉換微脂體-用於難以治療之癌症中央研究院生物醫學科學研究所羅傅倫特聘研究員發明人:羅傅倫 博士、白宸睿 博士/Steve Roffler PhD, BURNOUF Pierre-Alain PhD領域:新穎藥物適應症:胰臟癌/Pancreatic Cancer研發階段:候選藥物/醫材雛型試製造 Pilot production of candidate drug/prototype摘要:本研究目標為解決難以治療癌症的醫療迫切需求。微脂體是一種用於癌症治療的奈米技術,其發展之主要障礙包括藥物不易穩定滯留於載體內,不同藥物各別需要其專一且不同的裝載技術也不易同時包覆不同藥物在同一微脂體內。
我們開發的平台技術「醣苷轉換微脂體」利用化學修飾抗癌藥物上的醣苷轉換官能基以控制藥物在親脂的狀態下有效地負載於微脂體內,且以親水的狀態穩定滯留在微脂體內。「醣苷轉換微脂體」亦可安全穩定地被輸送至腫瘤部位,透過溶酶體內的葡萄醣醛酸酶水解其醣苷轉換官能基以產生具有活性之抗癌藥物並達到細胞毒殺效果。「醣苷轉換微脂體」的技術優勢為抗癌藥物滯留於微脂體內的穩定性及高承載力並可更進一步拓展其運用範疇於多重藥物的負載,由此我們可創造具協同效果之奈米藥物於癌症治療。This research seeks to address pressing medical needs associated with challenging-to-treat cancers. Liposomes are an established nanotechnology for cancer therapy, the major obstacles to the widespread development of liposomal nanomedicines include difficulties in stably retaining potent drugs inside the nanocarriers, identifying specific and unique conditions to load each drug, and problems with encapsulating different anti-cancer drugs in the same liposome.
Our platform technology, called glycosidic switch liposomes (GSL), can create highly potent and effective anti-cancer medications. This is achieved by chemically attaching a “glycosidic switch” to anti-cancer drugs. The glycosidic switch can be controllably interconverted between a lipid-soluble ester form for loading and a water-soluble glucuronide form for stable drug retention in the liposome aqueous core. GSL is stable in circulation and can be safely delivered to tumors for cellular uptake. The lysosomal enzyme beta-glucuronidase hydrolyzed the glycosidic switch to regenerate the parental drug inside cancer cells, resulting in cytotoxicity. The key advantages of GSL include the ability to securely retain potent anti-cancer drugs inside liposomes, achieve high drug loading efficiency, and serve as a versatile platform applicable to creating liposomal formulations of numerous drugs and drug combinations. Therefore, we are able to create a potent, high-payload,
and synergistic nanomedicine for cancer treatment.-
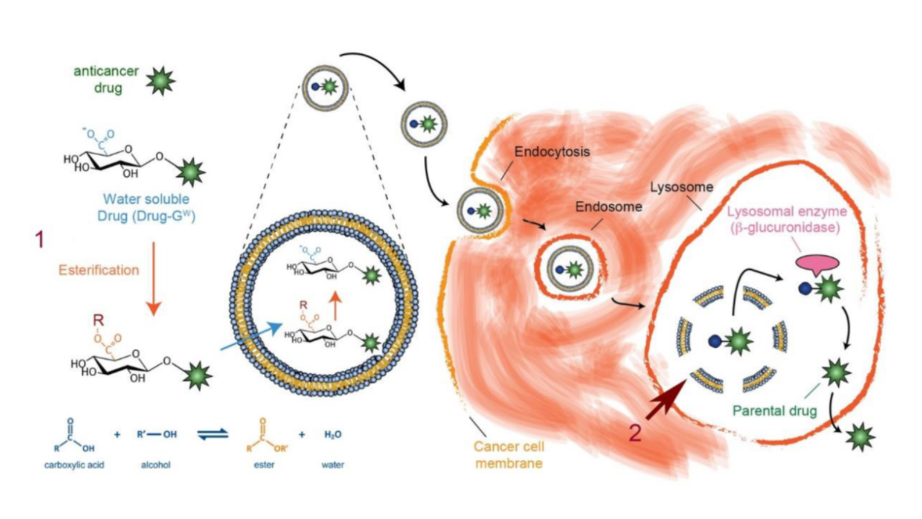
Schematic diagram of glycosidic switch strategy for drug loading in liposomes and intracellular drug regeneration:
1. A water-soluble glycosidic switch (GW) is attached to
a hydrophobic drug to generate a water-soluble drug (Drug-GW). The glycosidic switch can be esterified to produce a lipid-soluble form (Drug-GL) to facilitate active loading in
liposomes. At a high internal pH in the aqueous lumen of the liposomes, the glycosidic switch is spontaneously saponified to the water-soluble form, resulting in strong
retention of Drug-GW. Drug-GW liposomes are stable in circulation and can be safely delivered to tumors for cellular uptake. The glycosidic switch on Drug-GW is enzymatically
removed by the lysosomal enzyme beta-glucuronidase to regenerate the parental drug inside cancer cells.37New combination drug for pancreatic cancer therapy輔仁大學醫學系李紹禎副教授發明人:李紹禎領域:新穎藥物適應症:胰臟癌研發階段:動物驗證 In vivo validation摘要:胰臟癌雖然是去年國人癌症十大死因中的第七名,但是開發出針對它的新治療方法,目前仍具有為滿足醫療需求的迫切性。其原因主要是它的早期診斷不容易,一旦發現胰臟癌的出現大都已經是晚期,導致對它的治療不易,預後也不佳。
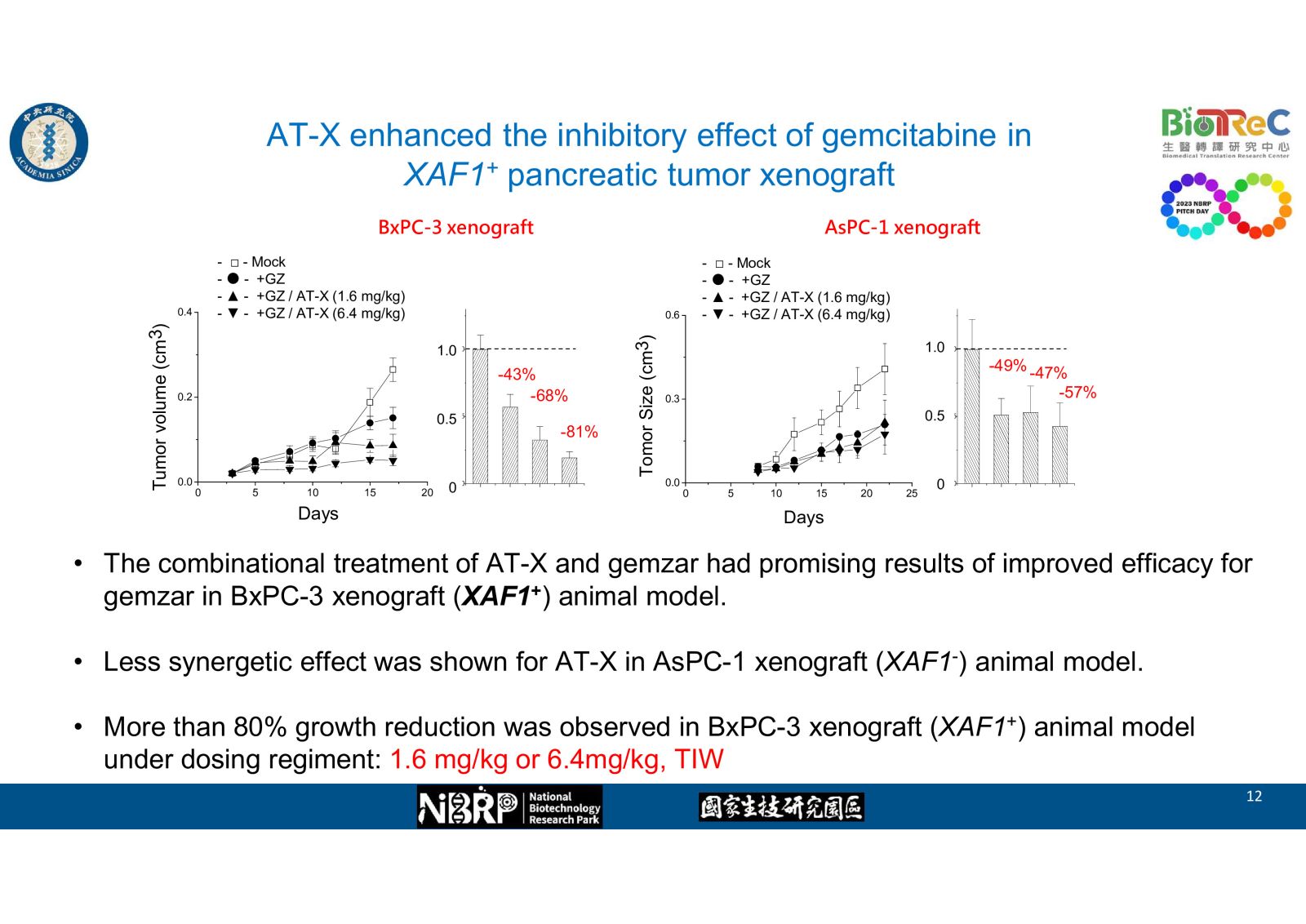
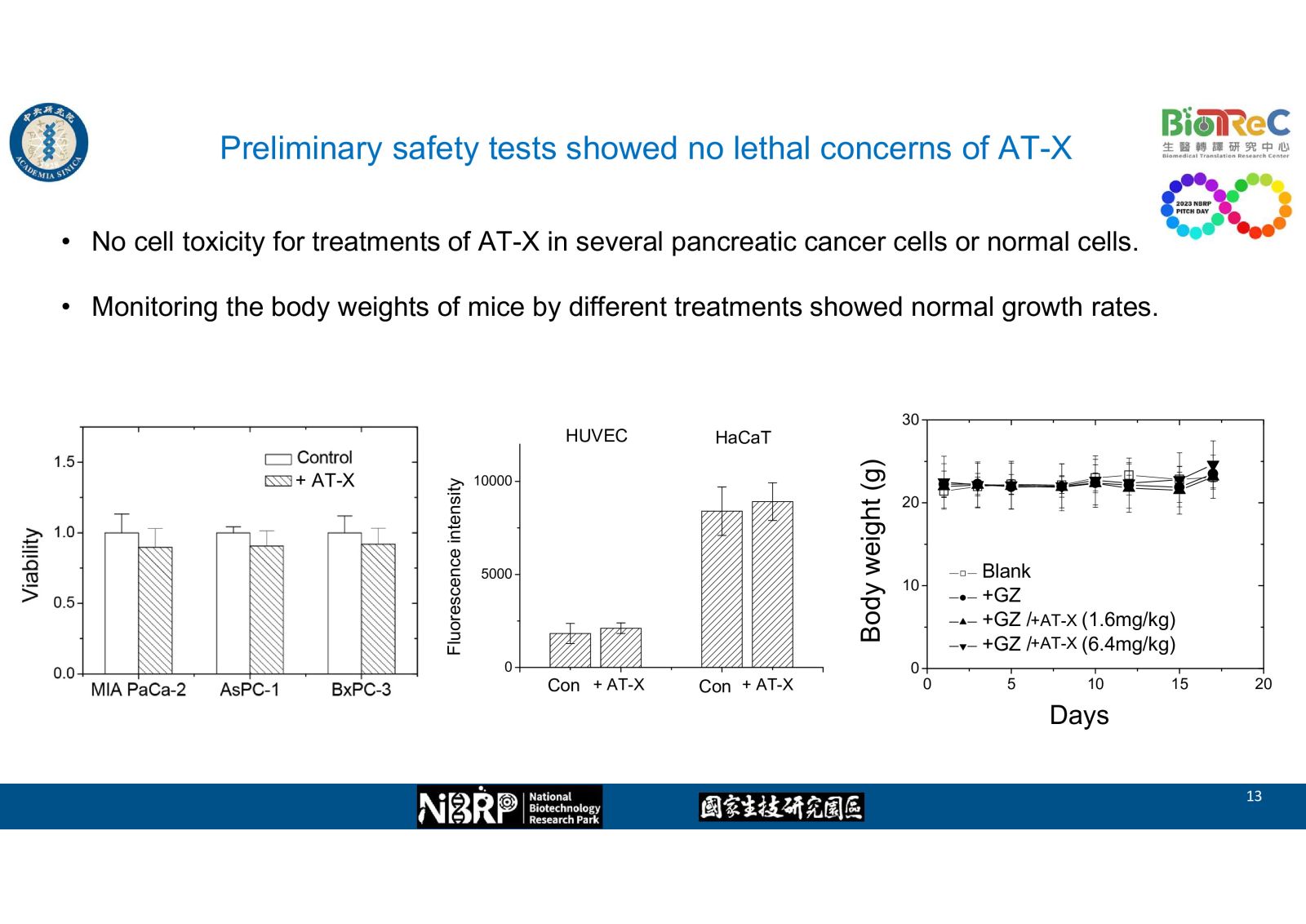
根據本實驗室的先前研究,我們發現了一個胰臟癌抗藥性的新分子機制: 當癌細胞受到化療藥物損傷後,細胞內的一個特定微核醣核酸miR-X 的增加,會減緩藥物作用造成的損傷,而導致抗藥性。藉此我們開發了一種抑制核酸,AT-X。在細胞實驗與動物實驗中,已證實了它的合併使用可以增加化療藥物Gemcitabine的敏感性,並且抑制實驗動物的皮下腫瘤生長。期望這能夠應用在治療胰臟癌,成為增加胰臟癌患者存活的新醫療策略。In 2022, pancreatic cancer was ranked as the 7th lethal neoplasia in Taiwan. There still exit unmet medical needs for its treatment. For its difficulty in early diagnosis, it was often confirmed as late stage carcinoma with poor response rate, poor prognosis. high recurrence, and less effective to current chemoagents
In our preliminary studies, we found on novel molecular mechanism of drug resistance in pancreatic cancer: upon cell damage by chemoagents, the level of one specific microRNA-X was elevated and thus the drug-induced damage was alleviated, which led to chemoresistance. Accordingly, we developed one nucleotide antagonist AT-X and demonstrated its effects at enhancing drug sensitivity and suppressed subcutaneous tumor growth in cell and animal studies. We hope this new drug combination coule be used in pancreatic cancer treatment and extend the patient survival.-
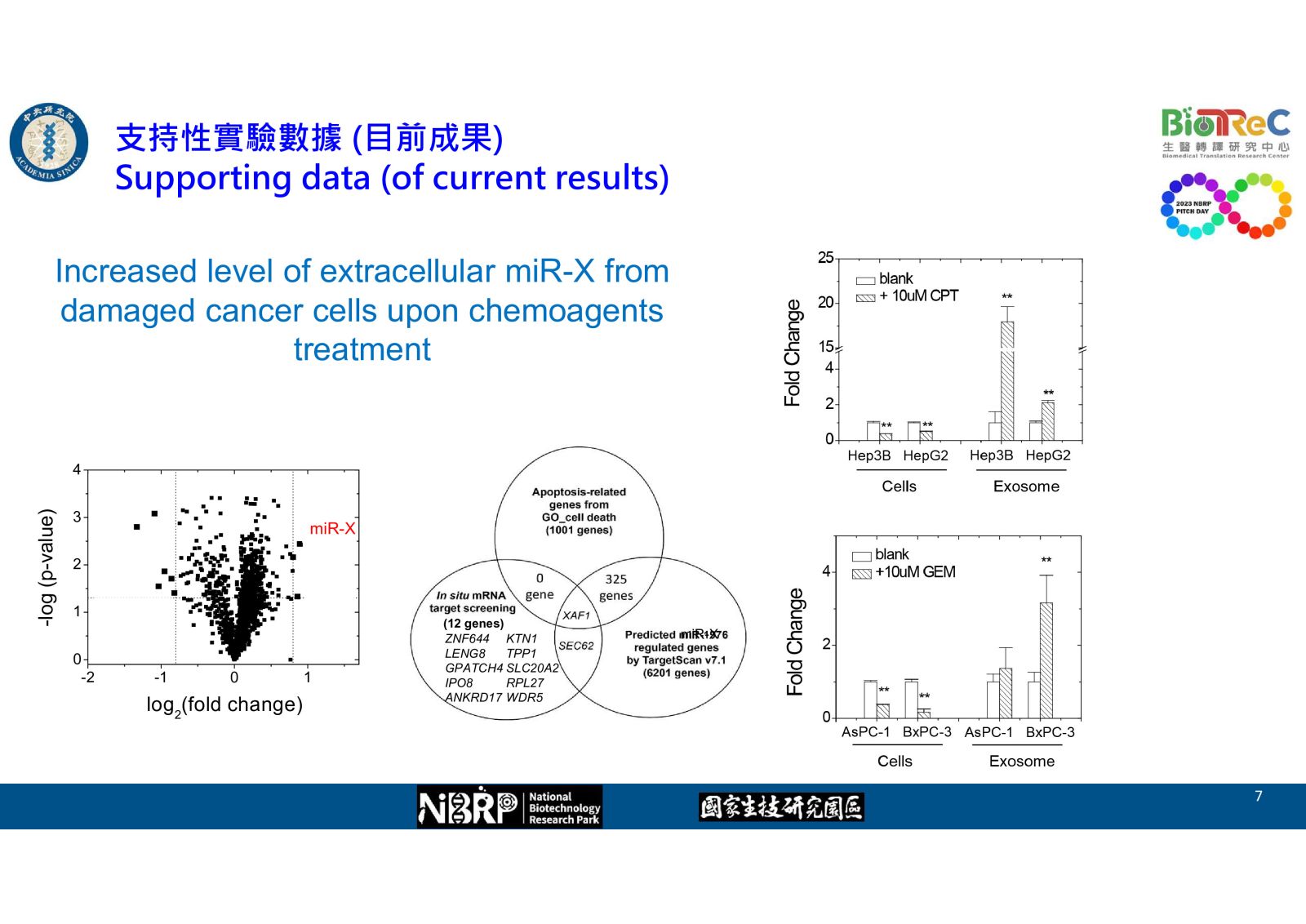
-- Increased level of extracellular miR-X from damaged cancer cells upon chemoagents treatment
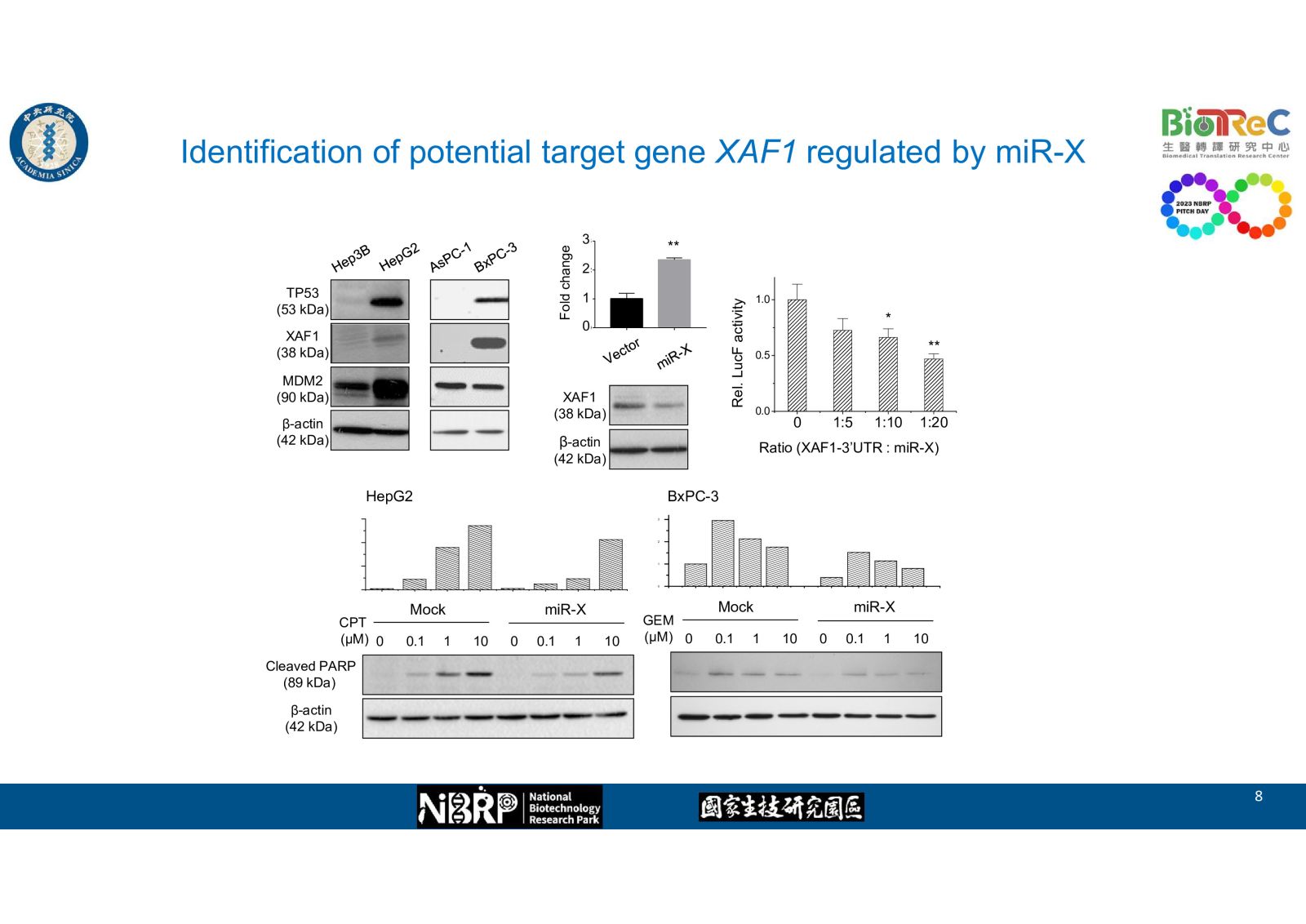
--Identification of potential target gene XAF1 regulated by miR-X
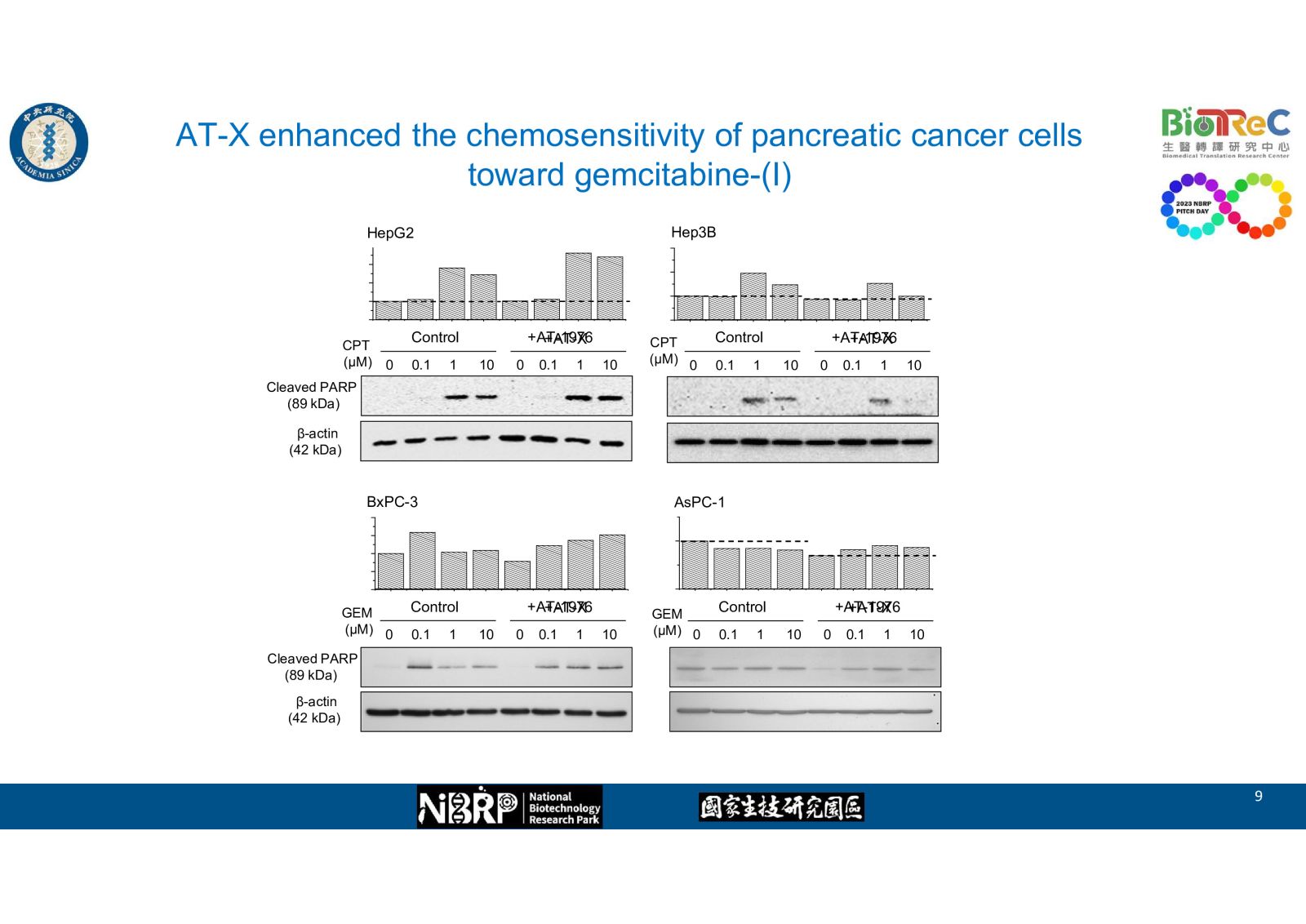
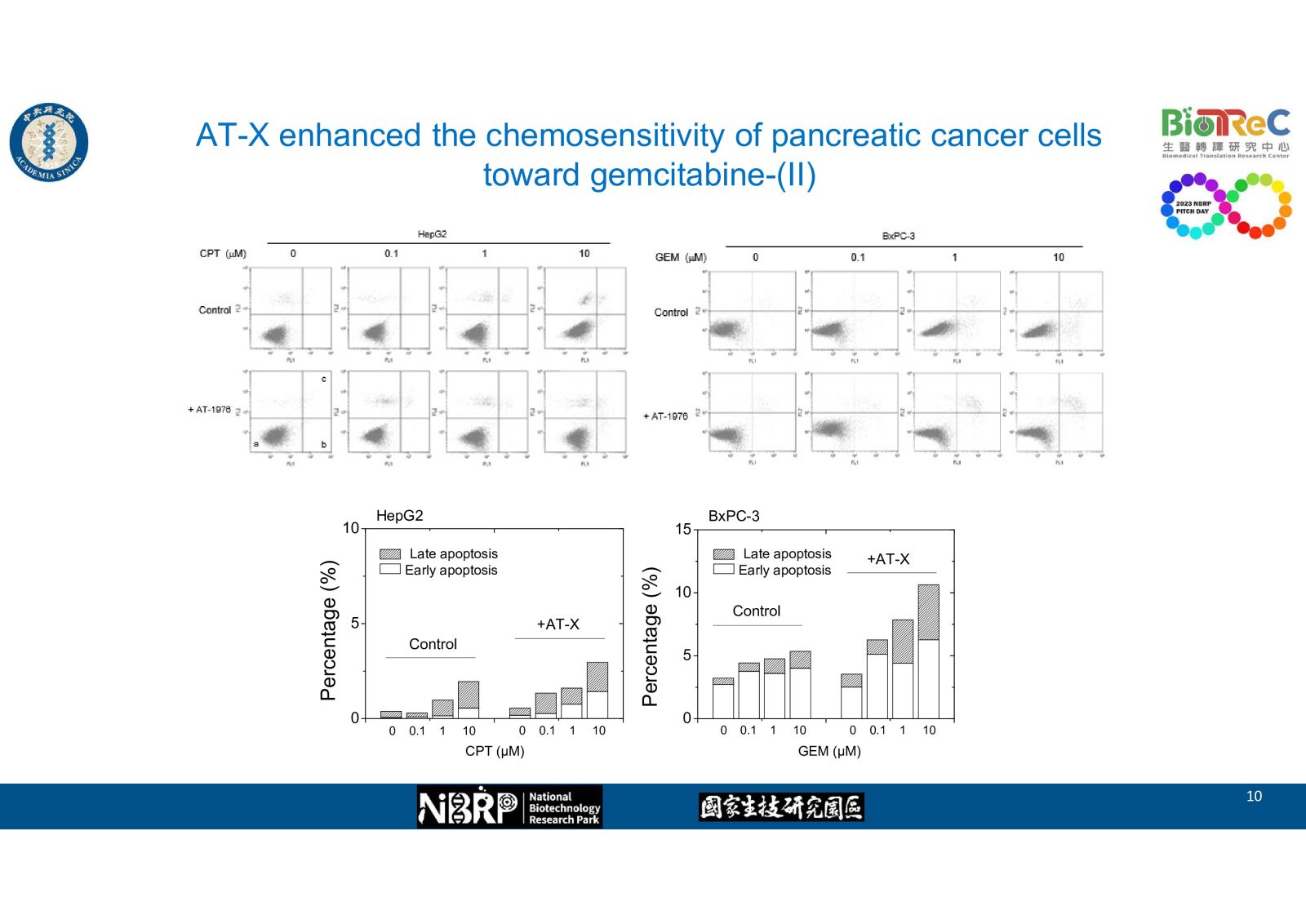
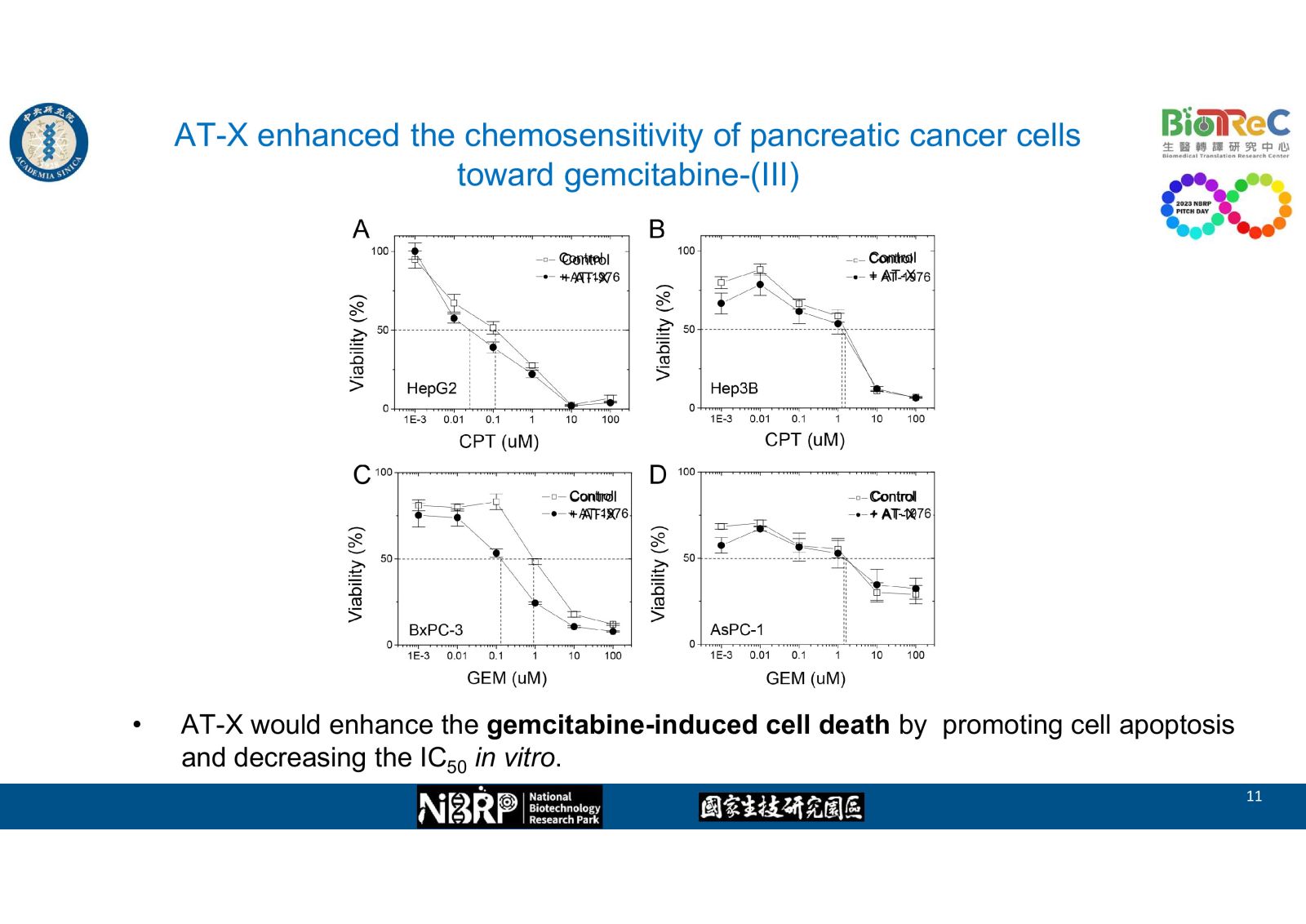
-- AT-X enhanced the chemosensitivity of pancreatic cancer cells toward gemcitabine as evidence by western blot, flow cytometry, and cell viability
-- AT-X enhanced the inhibitory effect of gemcitabine in XAF1+ pancreatic tumor xenograft
-- Preliminary safety tests showed no lethal concerns of AT-X38一項新穎核酸結構免疫刺激組合物在疫苗佐劑及腫瘤免疫治療上的應用國家衛生研究院/免疫醫學中心莊宗顯研究員發明人:莊宗顯、楊景行、曾仁志、伊曼娜、黃明熙、余冠儀領域:新穎藥物, 劑型開發適應症:疫苗佐劑及腫瘤免疫治療研發階段:動物驗證 In vivo validation摘要:一項新穎核酸結構免疫刺激組合物在疫苗佐劑及腫瘤免疫治療上的應用
莊宗顯
國家衛生研究院/免疫醫學中心
合成的CpG-寡脫氧核苷酸(CpG-ODN)是類鐸受體9 (TLR9) 激活劑。我們先前開發了一種新型 CpG-ODN,稱為 CpG-2722,它在誘導白細胞介素 12 (IL-12) 和干擾素 (IFN) 以及其他促炎細胞因子的產生方面具有良好的刺激活性。CpG-2722和抗PD-1單獨用於治療小鼠腫瘤時具有抗腫瘤活性,當二者合併使用時,抗腫瘤效果進一步增強。
環狀二核苷酸 (CDN) 是 STING 激活劑。 CpG-2722 和 CDN 均有核酸相關結構。 我們最近發現CpG-2722和不同CDN的組合物比它們單獨使用時具有更有效的免疫刺激活性。當 CpG-2722 和 2'3'-c-di-AM(PS)2 單獨用作疫苗佐劑時,可以增強對 SARS-CoV2 RBD 疫苗的免疫反應,但CpG-2722和2'3'-c-di-AM(PS)2合併使用時,佐劑作用進一步增強,使得SARS-CoV2疫苗更有效。 進一步的研究還顯示,即使流感病毒樣顆粒(VLP)本身已經具有強大的免疫刺激活,CpG-2722 與 2'3'-c-di-AM(PS)2 或 c-di-AMP 的組合物仍可有效的增強VLP疫苗的抗體反應。當用於動物模型中的癌症免疫治療時,CpG-2722/c-di-AMP組合物在腫瘤抑制方面比單獨使用CpG-2722更有效。這些揭示了這項新穎核酸結構免疫刺激組合物在疫苗佐劑和腫瘤免疫治療應用上的活性。
Application of a novel nucleic acid structure immunostimulatory composition in vaccine adjuvant and tumor immunotherapy
Tsung-Hsien Chuang
Immunology Research Center, National Health Research Institutes,
Zhunan, Miaoli, Taiwan
Synthetic CpG-oligodeoxynucleotides (CpG-ODNs) are activators of Toll-like receptor 9. We previously developed a novel CpG-ODN, called CpG-2722, which has good stimulatory activity in inducing production of interleukin-12 (IL-12),interferon (IFN), and other proinflammatory cytokines. The IL-12 and IFNs play a key role in linking innate and adaptive immunity together to elicit anti-tumor and anti-infectious immune responses. Therefore, we investigated the anti-tumor function of the CpG-2722. Both CpG-2722 and anti-PD-1 have anti-tumor activities when used alone in treatment of tumor bearing mice, the anti-tumor effect was further enhanced when the CpG-2722 and anti-PD-1 were used in combine.
Cyclic di-nucleotides (CDNs) are STING stimulators. CpG-2722 and CDNs contain nucleic acid related structures. We recently found that compositions of CpG-2722 and different CDN have more potent immune stimulatory activities that the use of CpG-2722 and the CDN alone. CpG-2722 and 2'3'-c-di-AM(PS)2 can boost immune responses to a SARS-CoV2 RBD vaccine when they were used alone as adjuvant for the vaccine. The adjuvant effect was further enhanced and making the SARS-CoV2 vaccine more effective when both of the CpG-2722 and 2'3'-c-di-AM(PS)2 were used in combine. Further studies, also revealed that compositions of CpG-2722 with 2'3'-c-di-AM(PS)2 or c-di-AMP effectively increase the antibody response of an influenza virus-like particle (VLP) vaccine, even the VLP itself already has potent immune stimulatory activity. When used for cancer immunotherapy in animal model, the CpG-2722/c-di-AMP composition was more effective in tumor suppression than the use of the CpG-2722 alone. These reveal the activity of the novel nucleic acid structure immunostimulatory compositions in vaccine adjuvant and tumor immunotherapy applications.-
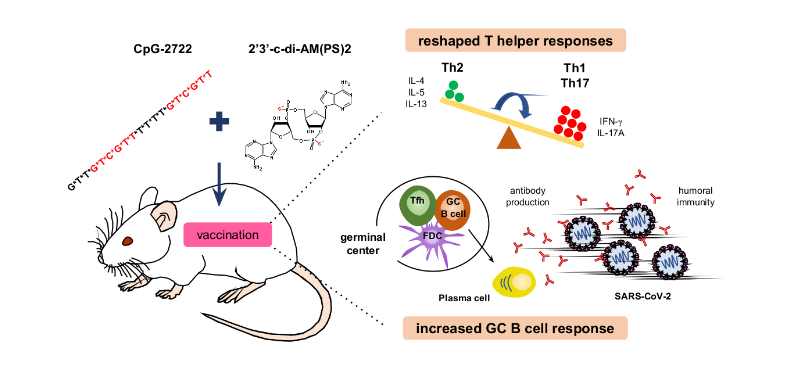
CpG-2722 和 STING 激活劑組合物的協同佐劑效應。 TLR9 激動劑CpG-2722 和 STING 激動劑 c-di-AM(PS)2 透過增強GC B 細胞反應和重塑 T 輔助細胞反應,協同增強對 SARS-CoV-2 RBD 疫苗的免疫反應。
39提高K他命用於治療神經精神疾患功效和安全性的新穎方法國家衛生研究院陳慧諴研究員發明人:陳慧諴領域:新穎藥物適應症:神經及精神疾病 (憂鬱症、複雜性疼痛、肌萎縮性脊髓側索硬化症、蕾特氏症、創傷後症候群等)研發階段:候選藥物/醫材雛型試製造 Pilot production of candidate drug/prototype摘要:低劑量K他命(ketamine)具快速抗憂鬱,改善自殺意念的發現為精神科藥物劃時代的突破。其後K他命陸續被證實對多種神經精神疾病也有顯著的療效。然而K他命的副作用和成癮性限制了其廣泛應用。本發明透過甜菜鹼與K他命併用,降低K他副作用和成癮性,並增進其療效,大幅提高K他命用於治療難治型憂鬱症、難治型躁鬱症、急性自殺意念發作等疾病的安全性。此技術已取得5國專利,全球化專利布局在全世界類似藥物之發展產業中居於領導地位。
The discovery of rapid antidepressant effects and improvement in suicidal ideation with low-dose ketamine marks a groundbreaking era in psychiatric medication. Subsequently, ketamine has been confirmed to have significant therapeutic effects on various neuro-psychiatric disorders. However, the side effects and addictive nature of ketamine limit its widespread application. This invention involves the combination of betaine with ketamine to reduce its side effects and addictive potential while enhancing its therapeutic effects, significantly improving the safety of ketamine in treating treatment-resistant depression, treatment-resistant bipolar disorder, acute suicidal ideation, and other conditions. This technology has been granted patents in five countries and holds a leading position in global patent layout within the industry of similar drug development worldwide.
-
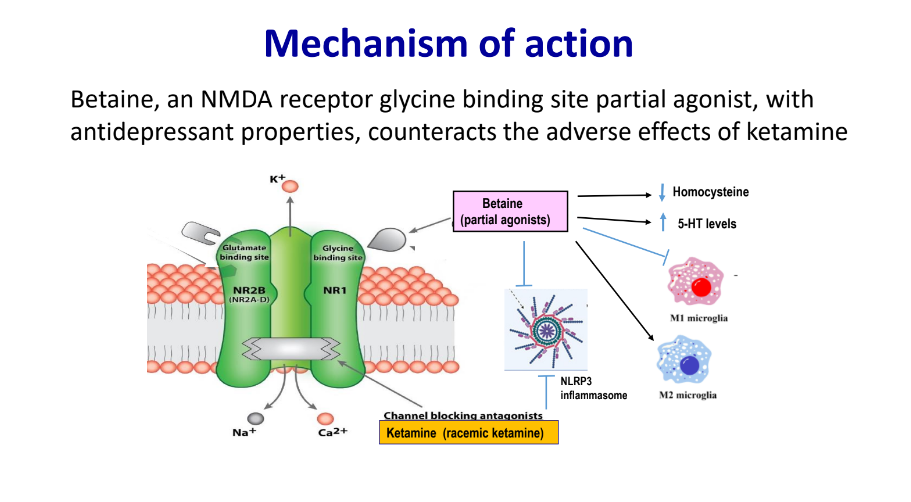
Betaine anhydrous, generally recognized as safe, is clinically used to treat homocystinuria. Betaine alone demonstrates remarkable antidepressant and analgesic effects. Betaine increases serotonin levels, inhibits NLRP3 inflammasome hyperactivation, and regulates microglial M1/M2 phenotypic differentiation, contributing to its antidepressant effects. Our team discovered that betaine also acts as a partial agonist at NMDA receptor glycine binding site, countering the adverse effects and addictive properties of ketamine.
40抗菌胜肽陰道凝膠治療細菌性陰道炎中央研究院細胞與個體生物學研究所臨海研究站陳志毅研究員發明人:陳志毅、林玟君領域:新穎藥物適應症:細菌性陰道炎研發階段:先導藥物最佳化 Lead drug optimization摘要:細菌性陰道炎好發於生育年齡的婦女,平均發生率約為三成。現階段抗生素治療伴隨著高復發率,導致病患反覆受疾病所擾,但臨床端的醫師卻只能重複開立老藥。
傳統抗生素治療高復發率主因有以下三點:
1. 無法根除生物膜,藥物無法有效達到作用標的,進而產生抗藥性菌株。
2. 目前使用之抗生素標的厭氧菌株,其演化已產生對現階段抗生素具有抗性之分支,導致一線抗生素治療為無效治療。
3. 細菌性陰道炎經常非由單一種厭氧菌之混合型感染,而目前抗生素治療僅針對抗厭氧菌,導致治療後其他非厭氧菌之增長。如:其副作用為陰道念珠菌感染。
為降低現今抗生素治療復發率,本技術為針對細菌性陰道炎,開發出具治療潛力之抗菌組合,已有多項數據證實優於傳統抗生素之特性。
Bacterial vaginosis (BV) is a common condition in reproductive-aged women, with a worldwide prevalence of over 30%. Current first-line antibiotic regimens are associated with a high recurrence rate for BV that may be due to an inability of antibiotics to effectively eradicate bacterial biofilms. The current first-line antibiotics, metronidazole and clindamycin, cannot inhibit many BV-associated bacteria. G. vaginalis and BV-associated bacteria often form a complex polymicrobial bio-structure in the BV biofilm, which is not only composed of anaerobic bacteria. It has been found that some aerobic bacteria, such as Streptococcus spp. and Escherichia coli, are also involved in BV pathogenesis. After the patient receives antibiotic treatment, co-existing microbes that are not sensitive to the antibiotics can become dominant species within the human vaginal microbiota. For example, the side effect of antibiotic treatment is vaginal candidiasis. When patients suffer from vaginitis repeatedly, they are more likely to experience sexually transmitted infections (STI), such as trichomoniasis or human immunodeficiency virus (HIV), making it difficult to restore a healthy environment. Therefore, new treatment strategies are needed for BV treatment. In order to improve treatment efficacy for BV, this technology develops a bactericidal combination with therapeutic potential for BV.
-
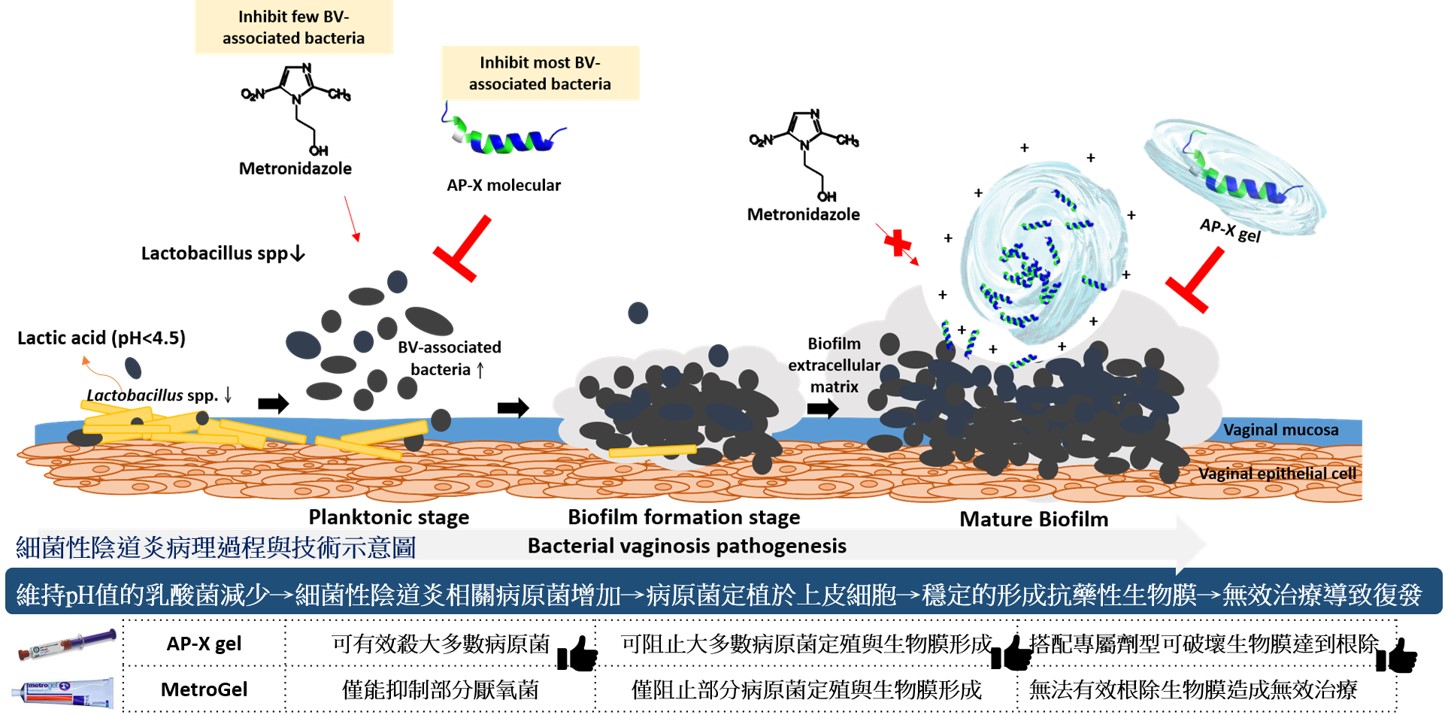
如圖